Здравствуйте, уважаемые коллеги!
Я начинающий специалист в области клинических исследований и хотел бы поддержать инициативу нового для меня сайта. Поскольку я только в начале своего профессионального пути и интересуюсь всем, что связано с клиническими исследованиями, то не могу описать свой пока небогатый опыт работы, но я часто читаю зарубежные материалы и так или иначе мысленно перевожу их, а в правилах сайта переводы не запрещены, так что я решил выложить перевод слайдов с PDA, которые мне, как новичку, показались интересными и возможно будут интересны другим новичкам 🙂
Конечно есть одна оговорка, информация на слайдах активно пересекается с требованиями, рассчитанные на США и ЕС, но поскольку наши требования НПП по-сути являются “гармонизированной” копией, то если абстрагироваться от слова FDA, то можно применить и к нам 🙂 А также автор значительную часть доклада посвятил GLP.
Сами слайды можно найти по этой ссылке (40 слайдов). Также благодарю администратора за помощь в оформлении слайдов. Пишите свои комментарии!
Слайд 1
GLP в сравнении с GMP и в сравнении с GCP
Доминик Пифат (Dominique Pifat), кандидат наук (Ph.D.), магистр делового администрирования (MBA)
Консультационная группа по биологическим препаратам
Слайд 2
Распространенное заблуждение
| Фундаментальные исследования | Исследование заболевания | Поиск лекарственного препарата | Доклинические исследования | Клинические исследования фаз I, II, III | Производство включая фарм. субстанции (ФС) Лаборатории КК |
| ← Не регулируется → | GLP | GCP | GMP | ||
← 21 CFR часть 11 → | |||||
⇒ | |||||
Основаны на исследовании | Основано на процессе | ||||
Часть 11 применяется для компьютеров, которые используют в регламентируемых FDA зонах.
Слайд 3
Надлежащая лабораторная практика (GLP)
- Система качества, охватывающая организационный процесс и условия, при которых планируют, проводят, мониторят, ведут учет, архивируют и сообщают доклинические исследования по безопасности для здоровья и окружающей среды.
- GLP представляет собой нормативные требования, опубликованные в Своде федеральных постановлений США (CFR) (21 CFR, часть 58)
- Принципы Организации экономического сотрудничества и развития (ОЭСР) в GLP
- GLP не является руководством или рекомендацией, она носит законодательный характер.
- GLP не является «урезанной» GMP!
Слайд 4
Надлежащая производственная практика
- Надлежащая производственная практика является частью системы обеспечения качества, гарантирующей постоянство (стабильность) производства и контроля продукции по стандартам качества в соответствии с её предусмотренным применением и требованиями регистрационного удостоверения или спецификации продукта.
- GMP охватывает как контроль производства, так и контроль качества
- Правила, регулируют лекарственные препараты в ЕС; Том 4 – Руководства ЕС по GMP для медицинских и ветеринарных препаратов
Слайд 5
Надлежащая клиническая практика
- GCP является международным этическим и научным стандартом качества по планированию, проведению, учету и сообщению исследований с участием человека.
- Соблюдение требований данного стандарта предоставляет общественные гарантии того, что права, безопасность и благополучие субъектов исследования защищены, согласуются с принципами, изложенными в Хельсинкской декларации, и то, что клинические данные являются достоверными.
- Руководством по Надлежащей клинической практике является гармонизированное трехстороннее руководство Международной конференции по гармонизации (ICH)
Слайд 6
Нормативные требования FDA к GLP применимы к (58.1)
- Доклиническим лабораторным исследованиям, которые подтверждают или предназначены для подтверждения заявки на исследование или разрешения на реализацию следующих продуктов:
- пищевые продукты и красители
- медицинские и ветеринарные препараты
- медицинские изделия
- биологические препараты
- электронные продукты
- GLP не имеет никакого отношения к производству препарата!
Слайд 7
Доклинические лабораторные исследования (58.3)
«In vitro или in vivo эксперименты, в ходе которых испытуемые препараты исследуют проспективно на предмет их безопасности в испытательных системах в условиях лаборатории.
Данный термин не включает исследования с участием человека или клинические исследования, или полевые исследования на животных. Данный термин не включает фундаментальные поисковые исследования, выполняемые с целью определения того, обладает ли испытуемый препарат потенциальной полезностью или для определения его физических или химических характеристик.»
Слайд 8
Базовые элементы GLP
| Персонал | Документы |
|
|
| Рабочий объект | Испытуемые и контрольные препараты |
|
|
Слайд 9
Основные обязанности
| Функция | GLP | GMP | GCP |
| Ответственность | Руководство рабочего объекта | Производитель | Спонсор |
| Главный ответственный за деятельность | Руководитель исследования | Уполномоченное лицо | Главный исследователь |
| Ответственный за «Выполнение рабочих операций» | Главный исследователь | Начальник производства | Фармацевт |
| Качество | Служба обеспечения качества | Начальник службы обеспечения качества/контроля качества | Служба мониторинга/обеспечения качества |
| Архив | Архивариус | Архивариус |
Слайд 10
Спонсор (58.3)
Под «Спонсором» подразумевают:
- лицо, которое инициирует и поддерживает проведение доклинического лабораторного исследования, путем предоставления финансовых или иных ресурсов
- лицо, которое подает доклиническое исследование на рассмотрение в FDA в поддержку заявки на исследование или разрешения на продажу
- исследовательский центр, если он одновременно инициирует и фактически проводит исследование
Слайд 11
Обязанности руководства (58.31)
- Назначение Руководителя исследования до начала инициирования исследования
- Оперативная замена руководителя исследования, при необходимости
- Удостоверение в наличии Службы обеспечения качества
- Удостоверение в том, что испытуемый и контрольный препарат надлежащим образом испытаны на предмет подлинности, дозировки, чистоты, стабильности и однородности, соответственно
- Утверждение существенных изменений в СОП-ах
- Удостоверение в том, что любое отклонение от правил GLP, сообщенное службой обеспечения качества, передано руководителю исследования, а также приняты и задокументированы соответствующие корректирующие меры
Слайд 12
Руководитель исследования (58.33)
«Для каждого доклинического лабораторного исследования должен быть назначен Руководитель исследования, ученый или другой специалист с соответствующим образованием, подготовкой и опытом, или их сочетанием.»
Слайд 13
Руководители исследования
- Только одно лицо назначается в качестве Руководителя исследования
- Соруководитель исследования не может быть назначен
- Может быть назначен альтернативный Руководитель исследования
- Правила GMP не содержат никаких требований к Руководителю исследования
- Правила GCP требуют наличия Главного исследователя
Слайд 14
Службы обеспечения качества (58.35)
«Исследовательский центр должен иметь Службу обеспечения качества, которая несет ответственность за мониторинг каждого исследования для гарантии того, что управление рабочими объектами, оборудованием, персоналом, методиками, практиками, записями и контролями соответствует нормативным требованиям.
Для каждого конкретного исследования Служба обеспечения качества должна быть полностью отделена и независима от персонала, задействованного в управлении и проведении исследования.»
Слайд 15
Обязанности Службы обеспечения качества (58.35) (b)(1)
- Сопровождение копии основной ведомости-графика всех доклинических исследований, проведенных в исследовательском центре и упорядоченных по:
- испытуемому препарату
- испытательной системе
- характеру исследования
- дате начала исследования
- текущему состоянию исследования
- Спонсору
- Руководителю исследования
Слайд 16
Обязанности Службы обеспечения качества (продолжение)
- Сопровождение копий всех протоколов
- Инспектирование каждого доклинического исследования через соответствующие интервалы времени
- Сопровождение подписанных отчетов каждой инспекции
- дата инспекции
- проинспектированное исследование
- проинспектированная фаза
- лицо, выполняющее инспекцию
- полученные результаты
- рекомендации
Слайд 17
Общие обязанности СОК
Существует 3 вещи, за проверку которых отвечает Служба обеспечения качества:
- Проводится ли исследование в соответствии с протоколом
- Проводится ли исследование согласно соответствующих СОП
- Проводится ли исследование в соответствии с правилами GLP
Слайд 18
Роль СОК
- СОК пересматривает СОПы на соответствие требованиям GLP
- СОК не утверждает СОПы (в рамках GLP)
- Правила GLP не требуют, чтобы СОК подписывала СОПы (это промышленный стандарт)
- СОК отвечает за предоставление гарантий руководству о том, что рабочие процедуры соответствуют СОПам
- СОК отвечает за сообщение отклонений руководителям исследований и руководству
Слайд 19
Рабочие объекты
- Правила GLP не требуют валидации какого-либо рабочего объекта
- Они не содержат требования к классификации воздуха
- Они не содержат специфического требования к санитарной обработке помещения (in vitro исследования)
- Они не содержат специфического требования к водопроводно-канализационной сети, сварным швам…
Слайд 20
Требования к оборудованию
К каждой единице оборудования должны быть в наличии:
- журнал оборудования
- СОП по эксплуатации
- СОП по калибровке и техническому обслуживанию
Правила GLP не требуют формальных процессов IQ, OQ, PQ
Слайд 21
21 CFR, часть 58.81
«Исследовательский центр должен иметь стандартные операционные процедуры, письменно определяющие методики к доклиническому лабораторному исследованию, которые Руководство утвердило, как пригодные для обеспечения качества и целостности данных, полученных в ходе исследования.»
Слайд 22
21 CFR, часть 58.81
«Каждая лаборатория должна иметь в непосредственном доступе лабораторные руководства и стандартные операционные процедуры к выполняемым лабораторным методикам. Опубликованная литература может быть использована в качестве дополнения к стандартным операционным процедурам.»
«Архив стандартных операционных процедур и все предыдущие редакции, включая даты этих редакций, должны сохраняться.»
Слайд 23
Служба ОК и СОПы
- СОК не пишет все СОПы, а только те, которые непосредственно относятся к функциям СОК
- СОК пересматривает СОПы на соответствие требованиям GLP
- СОК не утверждает СОПы (в рамках GLP)
- Правила GLP не требуют, чтобы СОК подписывала СОПы
- СОК отвечает за предоставление руководству гарантий того, что рабочие процедуры соответствуют СОПам
- СОК отвечает за сообщение отклонений руководителям исследований и руководству
Слайд 24
Управление СОПами
| GLP | GMP | GCP |
|
|
|
Слайд 25
21 CFR, часть 58.120
«Каждое исследование должно иметь утвержденный письменный протокол, в котором четко указаны цели и все методики для проведения исследования».
Протоколы не должны содержать «пропусков для заполнения»; они являются самостоятельными готовыми документами.
В правилах GLP нет никаких протоколов на серию
ОЭСР: План исследования
Слайд 26
Неофициальная иерархия GLP
Правила GLP > Протокол > СОПы
- Правила GLP превалируют над протоколом и СОПами
- Протокол всегда превалирует над СОПами
Слайд 27
Поправки к протоколу
«Все изменения или редакции утвержденного протокола и их причины должны быть задокументированы, подписаны Руководителем исследования и должны сохранятся вместе с протоколом».
- Поправки к протоколу предназначены для документирования постоянных изменений
- Если отклонение от протокола является ошибкой, то оно не должно вести к внесению поправки к протоколу (правила GLP не требуют проведения расследования)
- По возможности, поправки к протоколу должны быть перспективными
- По возможности, поправка к протоколу должны быть распределена получателям до того, как предполагается осуществление изменения (что включает СОК)
Слайд 28
Заключительные отчеты: 58.185
Заключительный отчет должен быть подготовлен для каждого доклинического лабораторного исследования и должен включать, но не ограничиваться следующим:
- название и адрес рабочего объекта, проводящего исследование
- даты начала и завершения исследования
- дату подписания протокола Руководителем исследования
- дату подписания заключительного отчета Руководителем исследования
Слайд 29
Заключительные отчеты: 58.185
Описание всех обстоятельств, которые могли повлиять на качество или целостность данных
- сообщают обо всех отклонениях от протокола и требований GLP
- затем Руководитель исследования принимает решение о том, влияют ли эти отклонения на качество или целостность данных
- Это выполняется в форме «Заявления о соблюдении требований»
- Заявление о соблюдении требований подписывается Руководителем исследования, а не СОК
Слайд 30
Заявление о соблюдении требований GLP
Протокол № 09802 исследовательского центра компании «Стерлинг Байотек» (Sterling Biotech)
Исследование безопасности на хомяках кандидата новой вакцины (R1009) против инфекционного дерматита промежности
Это исследование было проведено в исследовательском центре компании «Стерлинг Байотек» (SBRF) в соответствии с правилами надлежащей лабораторной практики Управления по продуктам питания и лекарственным препаратам США (21 CFR, часть 58) и применимыми стандартными операционными процедурами, за исключением случаев, указанных в отчете или исходных данных:
| Руководитель исследования: | Дата: |
Слайд 31
Заявление об обеспечении качества
Заявление об обеспечении качества
«Самый лучший исследовательский центр»
Название исследования:
Протокол №:
Инспекции по обеспечению качества и пересмотр вышеуказанного заключительного отчета по исследованию были проведены в соответствии с процедурами, описанными в стандартных операционных процедурах службы обеспечения качества и в соответствии с общими требованиями Правил надлежащей лабораторной практики, 21 CFR, часть 58, выпущенными FDA. Результаты инспекций и пересмотр заключительного отчета были сообщены Руководителю исследования и руководству в следующие сроки:
| Аудитор, выполнивший инспекцию/пересмотры | Результаты сообщены (дата) |
| – | – |
Слайд 32
Архивирование
- В условиях GLP, FDA имеет право и будет проверять ваш архив. Инспектора физически проверят то место, где хранятся ваши документы
- В условиях GMP, FDA не имеет четких требований к аудиту объектов, где хранятся архивы
Слайд 33
Ни одно из указанного на картинке ниже не относится к GLP
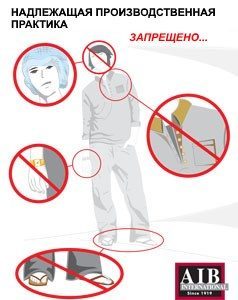
Слайд 34
Хранение записей: 21 CFR, часть 58.195, FDA
Документальные записи, исходные данные и образцы, относящиеся к доклиническому лабораторному исследованию, должны храниться в архиве (-ах), в течение того периода, который является более коротким:
- Как минимум два года с момента одобрения FDA заявки на исследование или разрешения на продажу, в поддержку которого были получены результаты доклинического лабораторного исследования
- Как минимум пять лет с момента подачи на рассмотрение в FDA результатов доклинического лабораторного исследования в поддержку заявки на исследование или разрешения на продажу
Слайд 35
Инспекции FDA
- В США нет способов, с помощью которых вы можете запросить инспекцию по GLP
- В США нет программы сертификации по GLP, подобной программам сертификации по ISO
- Подача данных на рассмотрение в регуляторные органы – это то, что вызывает инспекцию по GLP
Слайд 36
Четыре вида инспекций FDA
- Инспекция по GLP: периодическое рутинное определение соответствия лаборатории требованиям GLP (обычно включает проводимое исследование, а также завершенное исследование). Проводится по плану раз в два года.
- Аудит данных: инспекция, проводимая для проверки того, что информация, содержащаяся в заключительном отчете, поданном на рассмотрение в FDA, является точной и отражает исходные данные.
- Целенаправленная инспекция (или инспекция причины): инспекции, проводимые по различным веским основаниям (сомнительные данные в заключительном отчете, информация от заявителей жалоб и т. д.).
- Последующая инспекция: инспекция, иногда проводимая после инспекции по GLP, в ходе которой были выявлены неприемлемые практики и условия. Цель последующей инспекции заключается в обеспечении принятия надлежащих корректирующих мер.
Слайд 37
Возможные действия в отношении Вас со стороны FDA
- Предупредительное письмо (оно есть в Интернете: www.fda.gov/foi/warning.htm)
- Повторная инспекция
- Неформальная беседа
- Валидация доклинического исследования третьей стороной
- Дисквалификация рабочего объекта
- Судебное предписание / возбуждение уголовного дела
- Приостановка или аннулирование разрешения на продажу
- Прекращение срока действия нового исследуемого препарата
Слайд 38
Вывод
- Не существует такого понятия как «GLP партии»
- Правила GLP не применяются к какой-либо стадии производства
- Продукт, произведенный для токсикологических исследований, должен быть исследован на безопасность. Эти in vitro и in vivo исследования должны быть проведены в соответствии с требованиями GLP
- Не существует такого понятия как «GCP партии»
- Клинический материал должен быть исследован на безопасность и эффективность с участием людей. Эти исследования с участием людей должны проводиться в соответствии с требованиями GCP
Слайд 39
Ход GLP, GMP и GCP операций может перекрываться
Хронология разработки лекарственного препарата | |||||||||||||||||||
Новые составы и показания проходят через весь жизненный цикл разработки лекарственного препарата | |||||||||||||||||||
| Валидация целевого поиска | Определение круга потенциальных молекул-кандидатов | Оптимизация круга потенциальных молекул-кандидатов | Доклинические исследования | Клинические исследования фазы I | Клинические исследования фазы II | Клинические исследования фазы IIB | Клинические исследования фазы III | Клинические исследования фазы IV | Коммерциализация лекарственного препарата | ||||||||||
| Научный поиск и доклинические исследования | Ранние клинические исследования | Международные клинические исследования | |||||||||||||||||
| Клинические лаборатории | |||||||||||||||||||
| Биоанализ, биоаналитика | |||||||||||||||||||
Слайд 40
Столько людей работает в течение года для создания такого количества лекарственного препарата!








Уважаемый Максим, поддерживаю ваше желание вести обоснованные дискуссии!
Да, это можно перевести как
Не придираясь, отмечу что в оригинале отсутствует слово “System”, хотя не принципиально. Ведь речь о том (надеюсь, что следует понимать вас так), что ФСК состоит из GMP и риск-менеджемнта:
Уважаемый, Михаил, честно признаюсь, что некоторые слова в переводе добавил самостоятельно для улучшения восприятия самого перевода и с учетом других перечитанных мной текстов по GMP/GLP/GCP. Вы совершенно правы в том, что в оригинале нет слова система, но я так полагаю, что автор сознательно опускал некоторые слова, как в случае с СОК, он писал ОК, и учитывая ограниченное пространство на слайдах, вероятно, слушатели или читатели должны сами домыслить пропуски. Не первый раз вижу, как англоговорящие авторы делают сокращения, кажущиеся нелогичными для нас.
Спасибо автору!
Отмечу, что слайды содержат ряд ошибок. Некоторые исходят из ключевой фразы GMP:
1.2 GMP applies to the lifecycle stages from the manufacture of investigational medicinal products, technology transfer, commercial manufacturing through to product discontinuation. However the Pharmaceutical Quality System can extend to the pharmaceutical development lifecycle stage as described in ICH Q10, which while optional, should facilitate innovation and continual improvement and strengthen the link between pharmaceutical development and manufacturing activities. ICH Q10 is
reproduced in Part III of the Guide and can be used to supplement the contents of this chapter.
Иными словами, GMP – только часть Фармацевтической системы качества. Последняя может включать в себя и остальные надлежащие практики.
Добрый день, уважаемый Михаил. Большое спасибо за ремарку!
Насколько я понимаю на Слайде 4 указано:
Что подтверждается Вашими словами и приведенной цитатой из руководства ICH.
Пока читал Ваши слайды, Максим, решил поинтересоваться и для себя некоторыми вопросами GLP, и к своему интересу нашел целую книжку-руководство ВОЗ.
Спасибо за Ваш труд! Да и слайдер пришлось сделать под слайды 🙂 Так что все замечательно вышло.