Итак, уважаемые коллеги, продолжаем цикл статей по примесям.
Публикую перевод второй части, выполненный Optisemist-ом.
Ссылки на оригинальные статьи и другие части цикла:
Часть 2 (оригинал статьи). Переводы других частей цикла: Часть 1, Часть 3
Общественность и фармацевтическая промышленность стала уделять больше внимания примесям в лекарственных препаратах, о чем свидетельствует внимание к фармацевтическим примесям в книгах, журнальных статьях, а также национальных и международных руководствах (1-10). Примеси могут оказывать значительное влияние на здоровье вследствие возможных тератогенных, мутагенных или канцерогенных эффектов. Поэтому контроль и мониторинг примесей в ФС является критическим вопросом при разработке и производстве лекарственных препаратов.
В части I этой статьи, которая появилась в выпуске Pharmaceutical Technology от февраля 2012 года, обсуждались разные виды и источники примесей с описанием конкретных случаев (11). В этой статье, части II, обсуждаются хиральные, полиморфные и генотоксические примеси (12, 13). В части III, которая будет опубликована в выпуске Pharmaceutical Technology в апреле 2012 года, будут рассматриваться разные пути деградации ФС, примеси возникающие при взаимодействии ФС-вспомогательное вещество во время производства готовой лекарственной формы, примеси-метаболиты, разные аналитические методы измерения уровней примесей и средства контроля примесей.
Хиральные примеси
Примеси могут присутствовать в виде энантиомеров хиральных соединений. Между энантиомерами наблюдались различия в отношении фармакологического и токсикологического профилей при наличии хиральных примесей in vivo (14, 15). Значимость стереохимической чистоты можно проиллюстрировать на примере формотерола, селективного агониста β2-адренорецепторов (16). Это соединение содержит два хиральных центра. Начальные исследования показали, что стереоизомер с абсолютной конфигурацией (R, R) был активен как β2-агонист, а активность изменялась в зависимости от порядка в последовательности (R, R) > (R, S) > (S, S) > (S, R). Последующие исследования показали гораздо большую разницу для эудизмического соотношения R, R/S, S, которое увеличивалось от 50 до 850, когда примесь эутомера в диастереомере уменьшалась приблизительно с 1,5 % до < 0,1 % (17). Подобные примеры стереохимических изомеров имеются для стереоспецифических веществ, таких, как (S)-энантиомер α-метилдопы, пиценадол, (R)-сопромидин, (+)-(S)-апоморфин и сертралин (18–24).
Другим примером является асенапина малеат, антипсихотик, принадлежащий к классу дибензооксепинопирролов. На основании фармакологии рецепторов, считается, что его эффективность опосредована антагонистической активностью в отношении допаминовых (D)-2 рецепторов и серотониновых (5-HT)–2A рецепторов (25). Асенапин проявляет геометрическую изомерию и представляет собою рацемат (+) и (-) энантиомеров. Он демонстрирует сравнимую аффинность к связыванию, а это значит, что транс-асенапин проявляет более высокую аффинность к D4 рецепторам, чем (+)/cis-асенапин (26).
Поскольку были показаны различия в фармакологических и токсикологических профилях у хиральных примесей in vivo, то это требует тщательного мониторинга хиральных примесей. Хотя разработка хиральных лекарственных средств в виде отдельных стереоизомеров является предпочтительным подходом, следует обращать внимание на нежелательные стереоизомеры, которые могут присутствовать как примеси или продукты разложения в действующем веществе или готовом продукте или могут возникнуть при метаболизме в биологических системах. Хиральные примеси в образцах фармацевтических продуктов могут возникнуть как побочные продукты процесса синтеза или в результате инверсии хиральных центров или вследствие химической деградации, а также в результате того и другого. Подобно этому, инверсия хирального центра может произойти in vivo в результате метаболизма, химической деградации или обоих этих явлений.
Существуют руководства по разработке хиральных соединений, опубликованные регуляторными органами разных стран мира, но они бывают неконкретными и позволяют делать различные интерпретации. Задачи присутствующие при разработке хиральных соединений сложны и необходим координированный подход с участием разных групп разработчиков. Мультидисциплинарный подход указывает направление разработки хиральных соединений путем скоординированных исследовательских усилий на разных фазах разработки (22–36).
Полиморфные примеси
Полиморфизм – это способность соединения существовать в более чем одной кристаллической форме, влияет на физические, химические и биологические свойства рассматриваемого соединения (37). Эти особенности могут оказать влияние на несколько характеристик фармацевтических систем, такие как показатели метаболизма, стабильность вещества и биодоступность. Регуляторными органами уделяется особое внимание к установленным полиморфным свойствам и их пониманию при регистрации новых лекарственных средств (38).
В руководстве Q6A Международной конференции по гармонизации (ICH), Спецификация: методики и нормы для новых фармацевтических субстанций и новых лекарственных препаратов: химические вещества, отмечено когда и как полиморфные формы следует контролировать и отслеживать (39). Из соображений стабильности в готовом продукте обычно используется самая стабильная форма. Однако метастабильные формы могут ненамеренно возникнуть под воздействием температуры, механической обработки и влаги при производстве или хранении готового продукта (40).
Загрязнение полиморфными примесями может негативно влиять на стабильность или фармакокинетические свойства готового продукта. Кроме того, FDA требует разработки валидированных методик для определения соотношения кристаллических форм в процессе хранения фармацевтической субстанции и готового продукта (41).
Например, оланзапин кристаллизуется в более чем 25 кристаллических формах, из которых форма II была признана наиболее стабильной и используется в готовых формах (42, 43). Оланзапин обесцвечивается на воздухе (44). Полиморфные формы I и II проявляют очень малое различие на своих диффрактограммах. Поэтому определение наличия формы I в форме II становится очень важным.
Известно, что сальметерола ксенафоат существует в двух кристаллических формах, из которых форма I является стабильной, а форма II является метастабильной полиморфной формой при комнатной температуре (45). Эти полиморфные формы были охарактеризованы при помощи дифференциальной сканирующей калориметрии, рентгеновской диффракции кристаллов, термогравиметрического анализа и обратнофазной газовой хроматографии (46). Коммерческий сальметерола ксенафоат представляет собой микронизированную форму с такой же кристаллической структурой, что и форма I. Однако коммерческая субстанция может содержать следовые количества полиморфной формы II, которая образуется в процессе микронизации.
Особенные случаи примесей
Когда разрабатываются новые процессы синтеза вещества, например, чтобы обойти патентные ограничения, эти процессы обычно начинаются с новых главных исходных веществ и включают другие промежуточные продукты, реагенты, растворители которые могут реагировать непредсказуемо, что приводит к появлению побочных продуктов и других синтетических примесей. Например, во время синтеза линезолида и пеметрекседа динатрия вследствие разного хода синтеза могут образовываться разные синтетические примеси.
Фармацевтические компании могут создавать новые процессы синтеза, которые различаются исходными веществами, растворителями, реагентами, условиями (например, температурой) и полиморфными формами. При использовании новых материалов или процессов синтеза, могут образоваться несколько примесей, которые не появлялись в основном или первоначальном синтезе ФС. Частные статьи Фармакопеи США, Европейской Фармакопеи, Британской Фармакопеи, Индийской Фармакопеи и Японской Фармакопеи могут не подойти для контроля тех примесей, которые образуются в результате другого процесса. После публикации частной фармакопейной статьи компании вынуждены изменять аналитические методики или контролировать такие примеси как нефармакопейные, включая генотоксические примеси, при помощи отдельных аналитических методов, таких как высокоэффективная жидкостная хроматография (ВЭЖХ) или газовая хроматография (ГХ).
Например, в ходе синтеза линезолида примеси, образующиеся из соединения бис-линезолида и бис-бензиловой примеси, образуются и в ходе исходного патентованного процесса (47–49). Некоторые опубликованные патенты имеют другие возможные синтетические примеси, которые невозможно разделить одной ВЭЖХ методикой и образуются в результате процессов синтеза отличных от процесса синтеза в основном патенте (47–55).
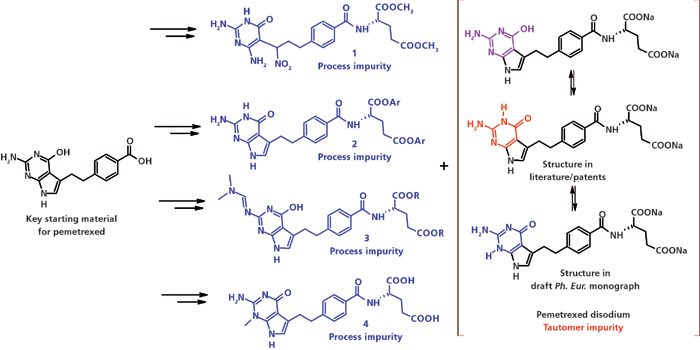 Рисунок 1: Реакционная схема для разных производственных подходов для примесей пеметрекседа натрия, обозначены соответственно как 1, 2, 3 и 4 (Лит. 57–60). Ph. Eur. – Европейская Фармакопея.
Рисунок 1: Реакционная схема для разных производственных подходов для примесей пеметрекседа натрия, обозначены соответственно как 1, 2, 3 и 4 (Лит. 57–60). Ph. Eur. – Европейская Фармакопея.
Пеметрекседа динатрия пентагидрат, ФС в препарате Алимта компании Эли Лилли, является многоцелевым антифолатом, применяемым для лечения мезотелиомы и во второй линии терапии при немелкоклеточном раке лёгких. Алимта также исследуется для применения при многочисленных других видах рака (56). Каждый, не вызывающий претензий патент, описывает разные возможные примеси (смотрите Рисунок 1, Примеси процесса синтеза 1, 2, 3 и 4) (57–60). Одна методика ВЭЖХ может оказаться непригодной для контроля всех этих примесей.
Примеси, производные от пиперазинового кольца
Пиперазиновая структура присутствует в молекулах более 200 лекарственных веществ. Биотрансформация пиперазинового кольца включает несколько хорошо известных метаболических реакций, включая N-окисление, гидроксилирование, N-деалкилирование и разрыв кольца до N-замещенных а также N,N’-дизамещенных этилендиаминов. Вдобавок сообщалось о нескольких неожиданных метаболических путях для пиперазинового кольца: N-глюкуронирование, N-сульфонирование, образование карбамоилглюкуронида и продуктов присоединения глутатиона (61). Некоторые соединения, содержащие пиперазиновое кольцо, показывают, что обычно это кольцо метаболически стабильно, когда оба атома азота замещены группами крупнее этила.
Отсутствие частичной деградации пиперазинового кольца в оланзапине (2-метил-4-(4-метил-1-пиперазинил)-10H-тиено[2,3-b][1,5]бензодиазепин) несколько удивляет. Были сообщения о некоторых основных метаболитах в человеческой плазме и моче, таких как 4′-N-глюкуронид (61, 62). Несколько других метаболитов были обнаружены в моче мышей, крыс, обезьян и собак (63). Однако об этилендиаминовой примеси не сообщалось ни как о метаболите, ни как о примеси процесса синтеза. (смотрите Рисунок 2).
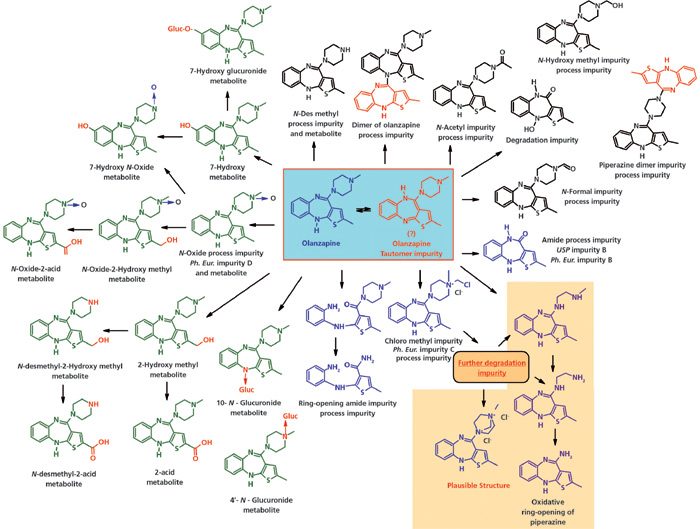 Рисунок 2: Пиперазиновое кольцо и примеси-метаболиты оланзанпина. Ph. Eur. – Европейская Фармакопея. USP – Фармакопея США.
Рисунок 2: Пиперазиновое кольцо и примеси-метаболиты оланзанпина. Ph. Eur. – Европейская Фармакопея. USP – Фармакопея США.
Когда в одном из атомов азота в пиперазиновом кольце водород замещается метиловой или этиловой группой обычно наблюдается образование этилендиамина. Примером является левофлоксацин, S-(-)-9-фторо-2,3-дигидро-3-метил-10-(4-метил-1-пиперазинил)-7-оксо-7H-пиридо[1,2,3-de][1,4]бензоксазин-6-карбоновая кислота, который является (S)-изомером офлоксацина. В левофлоксацине пиперазиновый атом азота замещается метильными группами с образованием нескольких примесей фотодеградации (смотрите от P 2 до P 10, Рисунок 3) (64–67). Наблюдаются также некоторые примеси процесса синтеза (смотрите Рисунок 3). Если в процессе синтеза левофлоксацина используют растворитель метилендихлорид, то может образовываться хлорометильная примесь, а после изоляции конечного продукта эта примесь может преобразоваться в дичетвертичную циклическую пиперазиновую примесь.
Также, когда в ципрофлоксацине (1-циклопропил-6-фторо-1,4-дигидро-4-оксо-7-(пиперазин-1-ил)хинолин-3-карбоновая кислота) в атоме азота в пиперазиновом кольце замещается водород, то образуется несколько метаболитов и примесей процесса синтеза (смотрите Рисунок 3) (68–74). Также наблюдалось, что когда во время синтеза происходит замещение гидрогена в азоте, то образуются две димерных примеси (F-F димер ципрофлоксацина и F-Cl димер ципрофлоксацина) (75).
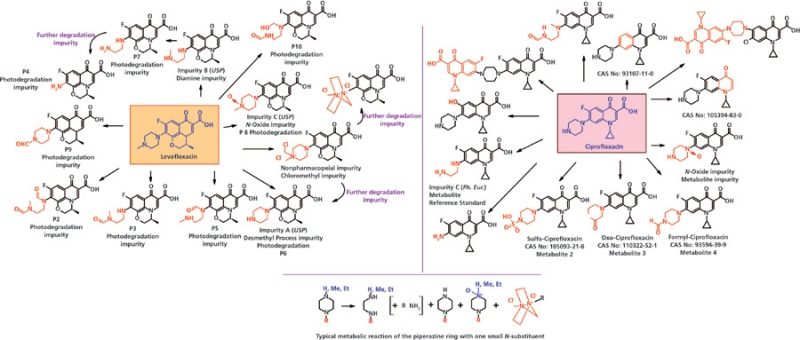 Рисунок 3: Пиперазиновое кольцо и примеси-метаболиты левофлоксацина и ципрофлоксацина. Ph. Eur. – Европейская Фармакопея и USP – Фармакопея США. CAS No. – номер Chemical Abstracts Service (CAS).
Рисунок 3: Пиперазиновое кольцо и примеси-метаболиты левофлоксацина и ципрофлоксацина. Ph. Eur. – Европейская Фармакопея и USP – Фармакопея США. CAS No. – номер Chemical Abstracts Service (CAS).
Генотоксические примеси
До 2000 года не было специальных документов о контроле генотоксических примесей. Руководства ICH упоминали мимоходом о соединениях с необычной токсичностью. Генотоксические примеси – это химические соединения, которые могут быть мутагенами и способны повреждать ДНК (76). Немоноалкилирующие агенты классифицируют как генотоксические вещества вследствие природы содержащихся функциональных групп, а также из-за связанных с ними аналиновых производных. Стадии преобразования в солевые формы также могут привести к образованию генотоксических примесей. Примерами являются образование метилхлорида, как побочная реакция при использовании соляной кислоты в метаноле, или образование эфиров метансульфоновой кислоты, как побочных продуктов стадии образования солей метансульфоновой кислоты, в спиртосодержащих растворителях. (77, 78).
EMA (Европейское агентство по лекарственным средствам) выпустило руководство о пороге токсикологической опасности (TTC, threshold of toxicological concern), которое рекомендует предел воздействия для потенциально генотоксических примесей – не более 1,5 мкг в сутки для коммерческих лекарственных средств (79). Согласно руководству требуется проведение испытания при помощи хорошо известного теста Эймса на сальмонеллах для всех потенциальных примесей процесса синтеза ФС, которые содержат структурные элементы, свидетельствующие о потенциальной генотоксичности. Тест Эймса – скрининговый тест, который помогает определить соединения, которые влияют на структуру ДНК. В тесте бактерии Salmonella подвергают воздействию соединения и определяют изменения в их росте. Эти изменения происходят при мутациях, когда структура ДНК изменяется в определенных местах, кроме того, также используют микроядерный тест (80, 81). Рекомендованные пороги квалификации, основанные на максимальной суточной дозе для фармацевтических субстанций и для готовых продуктов, предложены в руководствах ICH Q3A и ICH Q3B (7, 8). Для химиков-органиков, которые разрабатывают технологию синтеза ФС, была создана база данных о TTC (82). Руководство EMA рекомендует разработчикам процесса синтеза ФС избегать всех возможных ситуаций, которые могут привести к появлению в ФС любого количества примесей с генотоксических потенциалом.
При разработке методики контроля необходимо учитывать и другие факторы, такие как реакционная способность, растворимость, летучесть. Действия не должны основываться только на присутствии структур, которые вызывают опасение. Важно оценивать каждый случай отдельно, учитывать предшествующие данные – стадии, на которых образуются примеси, реакционную способность, степень переноса в ФС, разные пути введения, данные теста Эймса и данные о близких соединениях.
Во время разработки процесса синтеза генотоксические примеси могут быть введены как исходные материалы, реактивы, промежуточные продукты, катализаторы, побочные продукты, изомеры, продукты деградации (83). Алкилгалиды, которые используются как реагенты в синтезе, генотоксичны (84). Они также образуются во время химического синтеза, когда противоположный ион соли действующего вещества (гидрогенгалид) реагирует со спиртом, который используется как растворитель.
Генотоксины этилхлорид, метилхлорид и изопропилхлорид, образуются при получении соответствующих гидрохлоридных солей этанола, метанола и изопропилового спирта (растворители, включенные в список ICH) при низкой температуре (< 5 °C), что есть ключевым параметром для этих примесей. В спиртовых растворителях, когда используется 37 % водный раствор HCl или газообразная HCl, создается наибольшая вероятность образования алкилгалидных примесей в следовых количествах. Эти примеси обнаруживаются с помощью газовой хроматографии на уровне миллионных долей (ppm). Метансульфоновая кислота (мезилат), бензолсульфоновая (безилат) и p-толуолсульфоновая кислота (тозилат) широко используются как контр-ионы при образовании солей ФС (85–87). Взаимодействия этих кислот с остаточными количествами спиртов могут привести к образованию генотоксических примесей. Алкилметансульфонаты, алкилбензолсульфонаты и алкил-пара-толуолсульфонаты могут соединятся соответственно с иматиниба мезилатом, амлодипина безилатом, денаглиптина тозилатом (88, 89).
Внимание к генотоксическим примесям растет, что создает задачи как для химиков-синтетиков, так и химиков-аналитиков, по разработке чувствительных и эффективных методик обнаружения низких уровней примесей (то есть, ниже TTC < 1,5 мкг/сутки), что не всегда осуществимо и увеличивает время и стоимость разработки лекарственных средств.
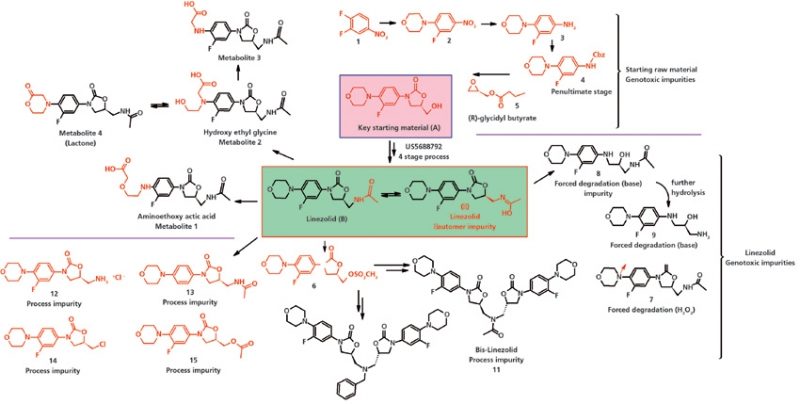 Рисунок 4: Технологические примеси, генотоксические и примеси метаболиты линезолида.
Рисунок 4: Технологические примеси, генотоксические и примеси метаболиты линезолида.
Линезолид, (S)-N-[[3-[3-фторо-4-(4-морфолинил)фенил]-2-оксо-5-оксазолидил]метил]-ацетамид, имеет структурные элементы, которые вызывают опасение о генотоксичности, а также представляет собою новый класс антибиотиков, оксазолидиноны. Исследования ускоренной деградации являются важной частью процесса разработки лекарственных средств и все больше используются при исследовании новых молекул. В этих исследованиях могут образовываться разнообразные примеси, которые могут не образоваться при оптимизации и валидации процесса производства, однако эти примеси подлежат контролю согласно руководств ICH. В исследованиях ускоренной деградации под воздействием пероксида водорода и в условиях щелочной среды авторы наблюдали образование примесей – Соединения 7, 8 и 9 (смотрите Рисунок 4), которые вызывали опасение в отношении генотоксичности и которые подлежат контролю на таком уровне, чтобы суточное воздействие не превысило 1,5 мкг/сутки с учетом максимальной суточной дозы линезолида.
Главное исходное вещество линезолида (А) вызывает опасение в отношении генотоксичности и оно включено в структуру 5 других промежуточных продуктов, Соединения 1, 2, 3, 4 и 5 (смотрите Рисунок 4). Соединение А преобразуется в фармацевтическую субстанцию и оно содержит мезил-примесь 6, амино-примесь 12, дезфторо- 13, хлор-примесь 14 и O-ацетил-примесь 15; всё это примеси процесса синтеза и они вызывают опасение в отношении генотоксичности (49). При исследовании на людях из введенного количества линезолида только 30 % выводилось через почки. Большая часть метаболизировалась путем окисления по морфолиновому кольцу, что приводило к образованию двух метаболитов (смотрите Рисунок 4): аминоэтоксиацетатного метаболитa и гидроксиэтилглицинового метаболита (то есть, это главные метаболиты в моче) (90–92).
Заключение
Часть II этой статьи включала обсуждение примесей, связанных с молекулами, содержащими один или более хиральных центров, ФС, существующими в разной кристаллической форме, ФС, содержащими пиперазиновый фрагмент, а также ФС, полученными по новым разработанным процессам синтеза. В части II также рассмотрено расширенное применение TTC к фармацевтическим продуктам. Для гарантии качества и безопасности лекарственных средств в процессе разработки предлагается концепция, включающая руководства ICH, которые акцентируют внимание на квалификации профилей примесей.
В части III, которая будет опубликована в выпуске Pharmaceutical Technology от апреля 2012 года, будут рассмотрены разные пути деградации ФС, примеси возникающие при взаимодействиях ФС-вспомогательное вещество при производстве готовой лекарственной формы, примеси-метаболиты, разные аналитические методики для определения уровней примесей и методики контроля примесей. В части I этой статьи, которая появилась в выпуске Pharmaceutical Technology от февраля 2012 года, обсуждались разные виды и источники примесей с описанием конкретных случаев (11).
Оставайтесь с нами и следите за появлением других частей цикла (от переводчиков ;))!
Литература
1. FDA, Guidance for Industry—ANDAs: Impurities in Drug Products (Rockville, MD, Aug. 2005).
2. FDA, Guidance for Industry—ANDAs: Impurities in Drug Substances (Rockville, MD, Jan. 2005).
3. S. Görög, Identification and Determination of Impurities in Drugs (Elsevier Science, Amsterdam, 2000).
4. S. Ahuja, Impurities Evaluation of Pharmaceuticals (Marcel Dekker, New York, 1998).
5. S. Hovorka and C. Schöneich, J. Pharm.Sci. 90 (3), 253–269 (2001).
6. J. Roy, AAAPS PharmSciTech3 (2), 1–8 (2002).
7. ICH, Q3A(R) Impurities in New Drug Substances (Feb. 2003).
8. ICH, Q3B(R) Impurities in Drug Products (Nov. 2003).
9. ICH, Q3C (R5) Impurities: Guideline for Residual Solvents (March 2011).
10. ICH, Q1A(R2) Stability Testing of New Drug Substances and Products (Nov. 2003).
11. K. R. Wadekar et al., Pharm. Technol. 36 (2), 46–51 (2012).
12. B.C. Allen, K.S Crump, and A.M. Shipp, Risk Anal.8 (4), 531–544 (1988).
13. S.W. Baertschi and D.W. Reynolds, “Introduction” in Pharmaceutical Stress Testing: Predicting Drug Degradation, J. Swarbick, Ed. (Taylor & Francis, New York, 2005), pp. 4–8.
14. S. Ahuja, Chiral Separations by Chromatography (Oxford University Press, New York, 2000).
15. S. Ahuja, Chiral Separations by Liquid Chromatography, ACS Symposium Series 471 (American Chemical Society, Washington, DC, 1991).
16. J. Trofast et al., Chirality3 (6), 443–450 (1991).
17. B. Waldeck, Chirality5 (5) 350–355 (1993).
18. L. Gillespie et al., Circulation25, 281–291 (1962).
19. H. Kubota et al., Chem. Pharm. Bull. 40, 1619–1622 (1992).
20. R.B. Carter, J. Pharmacol. Exp. Ther. 234 (2), 299–306 (1985).
21. Chiral Agonists of Histamine in Fornitier in Histamine Research (Oxford, 1985), pp.39-46.
22. M.E. Goldman et al., J. Mol. Pharmacol. 25 (1), 18–23 (1984).
23. W.M. Welch et al., J. Med. Chem. 27 (11), 1508–1515 (1984).
24. B.K. Koe et al., J. Pharmacol. Exp. Ther. 226 (3), 686–700 (1983).
25. T. de Boer et al., Chromatogr.26 (2), 156–165 (2012).
26. TGA, Australian Public Assessment Report for Asenapine (Woden, Australia, April 2011).
27. S.G. Allenmark, Chromatographic Enantioseparation: Methods and Applications (Ellis Horwood, Chichester, 1991).
28. G. Gubitz, Chromatographia30, 555–564 (1990).
29. D.E. Drayer, Clin. Pharmacol. Ther.40 (2), 125–133 (1986).
30. F.J. Jamali, J. Pharm. Sci. 78 (9), 695–715 (1989).
31. “FDA’s Policy Statement for the Development of New Stereoisomer Drugs,” Chirality4 (5), 338–340 (1992).
32. H. Wilson et al., J. Pharm. Biomed. Anal.11 (11–12), 1167–1173 (1993).
33. A.G. Rauws, Chirality 6 (2), 72–75 (1994).
34. M. Gross et al., Drug Inf. J. 27 (2), 53–457 (1993).
35. W.L. Heydorn, Pharm. News2, 19–21 (1995).
36. ICH, Q6A Document, Specification for New Drug Substances and Products: Chemical Substances, Step 2 version (1992).
37. J. Haleblian et al., J. Pharm. Sci. 58 (8) 911–929 (1969).
38. G.A. Stephenson et al., Adv. Drug. Deliv. Rev.48, 67–90 (2001).
39. ICH, Q6A Specification: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances (1999).
40. G.G. Z. Zhang et al., Adv. Drug Deliv. Rev.56 (3), 371–390 (2004).
41. S.R. Vippagunta et al., Adv. Drug Deliv. Rev.48 (1), 3–26 (2001).
42. P.B. Molinoff, The Pharmacological Basis of Theraputics (McGraw Hill, New York, 1996), pp. 399–430.
43. S.M. Reutzel-Edens, Cryst. Growth Des. 3 (6) 897–907 (2003).
44. Eli Lilly, “Olanzapine Polymorph Crystal Form,”US Patent 5,736,541 (April 1998).
45. H.Y. Tong et al., Pharm. Res. 18 (6), 852–858 (2001).
46. H.H. Tong et al., Pharm.Res.20 (9), 1423–1429 (2003).
47. D. Clemett et al., Drugs 59 (4), 815–827 (2000).
48. Teva Pharmaceutical Industries, “Isolated Bis-Linezolid, Preparation Thereof and Its Use as a Reference Standard, WO 2006/091848 A2, Aug. 2006.
49. Neuland Laboratories, “A Process for the Preparation of (5S)-(N)-[[3-[3-fluoro-4-4(4-morpholinly)phenyl-2-oxo-5-oxazolidinyl]methyl] acetamide,” WO2010084514A2, July 2010.
50. Symed Labs, “Novel Process for the Preparation of Linezolid and Related Compounds,” US Patent 2007/0032472, Feb, 2007.
51. B. A. Peralman, “Process to Produce Oxazolidiones,” US Patent 2002/0095054, July 2002.
52. Pfizer, “Process for Preparing Linezolid,” WO 2007/116284, Oct. 2007.
5 3. Pharmacia & Upjohn, “Process to Prepare Oxazolidinones,” US Patent 5837870, Nov. 1998.
54. Jubilant Life Sciences, “Process for the Preparation of Linezolid,” WO 2011/114241, Sept. 2011.
55. Pharmacia and Upjohn, “Substituted Oxazine and Thiazine Oxazolidinone Antimicrobials,” US Patent 5688792, Nov. 1997.
56. D.C.M Chan et al., Curr. Med. Chem.13 (4), 377–398 (2006).
57. Fortress Metro Hainan Tianyuan Pharmaceutical Technology Co., Ltd., “Nitro Compounds and the Preparation of Pemetrexed CN1827604A, Sept. 2006.
58. Wu Torrent, “Pemetrexed Intermediates and Preparation Methods, CN101085775A, Dec. 2007.
59. Dr. Reddy’s Laboratories, “Process for Preparing Pemetrexed, WO 2011/019986 A2, Feb. 2011.
60. D.P. Kjell et al., Org. Proc. Res. Dev.9 (6), 738–742, 2005.
61. K. Kassahun et al., Drug Metab. Dispos. 25 (1), 81–93 (1997).
62. K. Kassahun et al., Drug Metab. Dispos.26 (9), 848–855 (1998).
63. E. Mattiuz et al., Drug Metab. Dispos. 25 (1), 573–583(1997).
64. M. Lalitha Devi et al., J. Pharm. Biomed. Anal.50 (5), 710–717 (2009).
65. Y. Yoshida et al., Arzneimittelforschung43 (5), 601–606 (1993).
66. J. Sunderlanda et al., J. Antimicrob. Chemother.47 (3), 271–275 (2001).
67. A. V. Polishchuk et al., High Energy Chemistry42 (6), 459–463 (2008).
68. W. Gau, et al., Arzneimittel Forschung36 (10), 1545–1549 (1986).
69. P. Mojaverian et al., J. Pharm. Biomed. Anal.16 (3), 439–445 (1997).
70. A. Taicheng et al., Applied Catalysis B: Environmental94 (3–4), 288–294, 2010.
71. M. Mella et al., Helvetica Chimica Acta84 (8), 2508–2519 (2001).
72. K.A. Thabaj et al., Polyhedron26 (17), 4877–4885 (2007).
73. T.G. Vasconcelos et al., Chemosphere76 (4), 487–493 (2009).
74. K. Torniainen et al., J. Pharm. Biomed. Anal.15 (7), 887–894 (1997).
75. K. Tovarna Zdravil, “Process for Preparing Purified Ciprofloxacin,” WO 2005/075430, Aug. 2005.
76. R. J. Islam M et al., J. Pharm. Sci.90 (5), 541–544 (2001).
77. H.V Hogerzeil et al., British Medical Journal 304, 210–214 (1992).
78. R. J. Bhuiyan K et al., Indian Drugs34 (11), 634-636 (1997).
79. EMA, Guideline on the Limits of Genotoxic Impurities (London, June, 2006).
80. B.N. Ames and L.S. Gold, Proc. Nat. Acad. Sci. USA87 (19), 7772–7776 (1990).
81. B.N. Ames and L.S. Gold, Proc. Nat. Acad. Sci. USA 87 (19), 7777- 7781 (1990).
82. M.A. Cheeseman et al., Food Chain Toxicol.37, 387–412 (1999).
83. D.A. Pierson et al., Org. Proc. Res. Dev.13 (2), 285–291 (2009).
84. D.P. Elder. et al., J. Pharm. Biomed. Anal. 48 (3), 497–507 (2008).
85. D.P. Elder. et al., J. Pharm. Biomed. Anal. 46 (1), 1–8 (2008).
86. D.P. Elder. et al., J. Pharm. Sci. 99 (7,) 2948-2961 (2010).
87. G.E. Taylor. et al., J. Chromatogr A1119 (1–2), 231–237 (2006).
88. K. Ramakrishna. et al., J. Pharm. Biomed. Anal. 46 (4), 780–783 (2008).
89. N.V.V.S.S. Raman. et al., J. Pharm. Biomed. Anal.48 (1), 227–230 (2008).
90. J. G. Slatter et al., Drug Metab. Dispos.29 (8), 1136–1145 (2001).
91. N. Plock et al., Drug Metab. Dispos. l35 (10), 1816–1823 (2007).
92. Wynalda et al., Drug Metab. Dispos. 28 (9), 1014–1017 ( 2000).