
Уважаемые коллеги, продолжаем цикл статей по примесям.
Перевод третьей (последней) части, выполненный Ольгой Шуклиновой.
Ссылки на оригинальные статьи и другие части цикла:
Часть 3 (оригинал статьи). Переводы других частей цикла: Часть 1, Часть 2
Мониторинг и контроль примесей в ФС и готовых лекарственных средствах являются ключевыми вопросами в их разработке и производстве. В части I настоящей статьи, которая была опубликована в Pharmaceutical Technology в феврале 2012 года, обсуждались различные виды и источники примесей с рассмотрением конкретных случаев (1). В части II, опубликованной в мартовском выпуске 2012 года, рассматривались хиральные, полиморфные и генотоксические примеси (2). В части III, авторы рассматривают различные пути деградации ФС, примеси, возникающие вследствие взаимодействия ФС-вспомогательное вещество при производстве готовых лекарственных форм, примеси-метаболиты, разные аналитические методики для определения уровней примесей, а также способы контроля примесей в препаратах.
Определение примесей
Термин примесь обозначает нежелательные химические вещества, присутствующие в ФС или те, которые появляются в процессе изготовления лекарственной формы или при старении ФС в готовых лекарственных средствах. Наличие таких веществ даже в небольших количествах может повлиять на эффективность и безопасность препаратов. Несколько руководств Международной конференции по гармонизации (ICH) регламентируют примеси, присутствующие в новых субстанциях, лекарственных средствах, а также в остаточных растворителях (3-6). В соответствии с руководствами ICH по примесям в новых лекарственных средствах примеси, присутствующие в количествах ниже 0,1%, не должны считаться таковыми, за исключением, если для потенциальных примесей не прогнозируется необычайно сильное действие или токсичность (5). Во всех других случаях примеси должны быть определены как таковые. Если же количество примесей превышает пороговые значения, а данные, которые позволят подтвердить предложенный уровень спецификации, недоступны, то могут потребоваться исследования для получения таких данных. Несколько современных статей описывают разработанный подход и методические указания для выделения и идентификации примесей, которые возникают в процессе производства лекарственных средств (ЛС), а также продуктов распада с использованием масс-спектрометрии, спектроскопии ядерного магнитного резонанса (ЯМР), высокоэффективной жидкостной хроматографии (ВЭЖХ), а также ИК-Фурье спектроскопии (ИК-Фурье) фармацевтических субстанций (7-9).
Примеси – продукты распада
Продукты распада – это соединения, полученные вследствие разложения объекта исследования или действующего вещества. Некоторые примеси могут появляться в процессе распада ФС или других взаимодействий при хранении, поэтому для обеспечения безопасности препарата существует потребность в изучении его стабильности (10). Гидрохлортиазид (см. Рисунок 1) является классическим примером примеси – продукта распада.
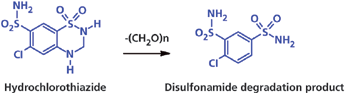
Рисунок 1: Деградация гидрохлортиазида.
Общеизвестен путь, по которому данное вещество распадается к исходному в его синтезе – дисульфамиду.
Продукты распада могут возникнуть в результате самого синтеза, хранения, изготовления лекарственной формы и старения (11). Вышеперечисленные пути распада обсуждаются далее.
Примеси, связанные с процессом синтеза.
Примеси в фармацевтических субстанциях или новых химических объектах образуются, главным образом, в процессе синтеза из исходных материалов, растворителей, промежуточных и побочных продуктов синтеза. Исходные материалы, как правило, производится с гораздо более низкими требованиями чистоты, чем фармацевтическая субстанция и, соответственно, абсолютно понятно, почему в них может содержаться множество компонентов, которые в свою очередь влияют на чистоту фармацевтической субстанции.
1-метил-3-фенил пиперазин (см. Рисунок 2) присутствует в виде непрореагировавшего исходного вещества, которое конкурирует на всех стадиях и в конечном итоге приводит к образованию примеси кетопиперазина – производного миртазапина (см. Примесь C, рисунок 2).
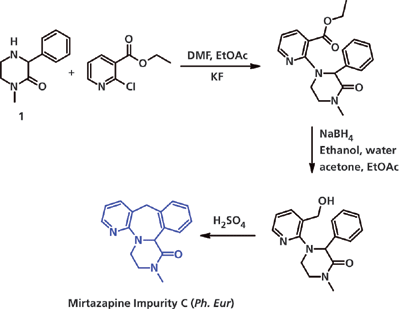 Рисунок 2: Реакционная схема примеси миртазапина. Ph. Eur – Европейская Фармакопея. DMF – диметилформамид. EtOAc – этилацетат.
Рисунок 2: Реакционная схема примеси миртазапина. Ph. Eur – Европейская Фармакопея. DMF – диметилформамид. EtOAc – этилацетат.
Примеси, связанные с производством лекарственного препарата.
Некоторые примеси в готовых ЛС или ФС могут образовываться вследствие взаимодействия со вспомогательными веществами, которые были использованы в процессе производства препарата. Во время производства ЛС, субстанция подвергается различным воздействиям, в результате которых возможны процессы распада или другие побочные реакции. Например, если во время высушивания или на других стадиях используется нагревание, то оно может способствовать распаду термически лабильных субстанций. В растворах и суспензиях распад вещества является более вероятным из-за процессов гидролиза или сольволиза. Эти реакции также могут встречаться при производстве твердых лекарственных форм, таких как таблетки и капсулы, когда вода или другой растворитель используется на стадии грануляции.
Есть два набора типичных условий при изучении реакций распада в твердом и жидком состояниях. Стандартными условиями для ФС в твердом состоянии могут быть
– 80 °С при относительной влажности 75% (ОВ),
– 60 °C при такой же влажности воздуха;
– 40 °С при относительной влажности 75% и световом излучении.
Типичными условиями для ФС в твердом состоянии могут быть:
– рН 1-9 в буферных средах, с перекисью и/или свободно-радикальными инициаторами;
– световое излучение.
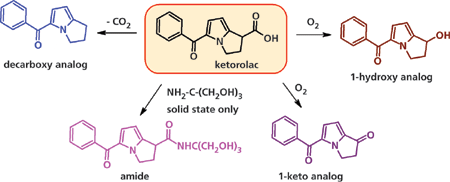 Рисунок 3: Путь деградации кеторолака.
Рисунок 3: Путь деградации кеторолака.
На рисунке 3 показан путь распада кеторолака в твердом и жидком состояниях (12-14).
Примеси, связанные с лекарственной формой.
Примеси, связанные с лекарственной формой, являются очень важными, поскольку многоразовое осаждение основного ингредиента требует изменения различных факторов, таких как рН или выщелачивания (15). Например, осаждение имипрамина гидрохлорида натрия бисульфитом требует последовательного изменения рН раствором лидокаина гидрохлорида в присутствии 5% раствора декстрозы в физиологическом растворе.
Примеси, связанные со способом производства.
Известная примесь 1-(2,6-дихлорфенил)индолин-2-он образуется непосредственно в ампулах натрия диклофенака. Образование этой примеси зависит от исходного рН препарата и условий стерилизации (например, в автоклаве при 123 °C ±2 °С), что приводит к межмолекулярной реакции циклизации натрия диклофенака в результате чего образуются производное индолинона и натрий гидроксид (16).
Экзогенные примеси.
Данные примеси могут возникать в результате воздействия следующих факторов:
- Температура. Многие термолабильные соединения теряют устойчивость при очень высоких температурах. Учитывая это, следует соблюдать предельную осторожность для предотвращения их распада.
- Свет (ультрафиолетовое излучение). Вследствие воздействия света происходят фотолитические реакции. Некоторые исследования показали, что эргометрин и его раствор для инъекций являются неустойчивыми в тропических условиях, при высокой инсоляции и температуре (17-19).
- Влажность. Влажность является одним из важных факторов при работе с гигроскопическими соединениями. Этот фактор может негативно влиять как на нерасфасованные порошки, так и на готовые твердые лекарственные формы. Общеизвестными являются примеры ранитидина и аспирина (19).
Примеси старения.
Как правило, чем длительнее препарат пребывает на полке, тем больше вероятность образования в нем примесей. Причиной появления таких примесей являются некоторые взаимодействия, которые описаны ниже.
Таблица 1. Последствия взаимодействия между компонентами
| Действующее вещество | Фармацевтическое вспомогательное вещество | Эффект |
|---|---|---|
| Канамицин | Подсластитель; сахарный сироп | Потеря активности при комнатной температуре |
| Холекальциферол | 2 % эфир полиоксиэтилена сурфактант; полисорбат | Изменение рН, распад действующего вещества |
| Тетрациклины | Кальций или магний или ионы металлов | Комплексообразование |
| Тиомерсал | Бром; соединения хлора; соединения йода | Образование различных расстворимых галидов или катионных соединений ртути |
| Адреналин | Борная кислота; повидон | Стабилизация |
Оригинал

- Взаимодействие между компонентами. Витамины очень неустойчивы в результате старения. Например, присутствие никотинамида в 4-х компонентной витаминной смеси (никотинамид, пиридоксин, рибофлавин и тиамин) вызывает распад тиамина к субстандартному уровню в течении первого года срока хранения (20). В таблице I приведены некоторые примеры взаимодействия между компонентами.
- Гидролиз. Многие препараты являются производными карбоновых кислот или содержат функциональные группы, которые чувствительные к кислотно-щелочному гидролизу (например, аспирин, атропин и хлорамфеникол).
- Окисление. В лекарственных средствах наиболее распространенной формой окислительного разложения является автоокисление через цепной, свободно-радикальный процесс. Препараты, которые склонны к окислению, включают метотрексат, адиназолам, катехоламины, сопряженные диены (например, витамин А), и нитрозо- и нитропроизводные. Оланзапин особенно поддается окислительному распаду в присутствии кислорода (Рисунок 4) (21).
- Фотолиз. Фотолитическое расщепление продуктов старения происходит с ФС или лекарственными препаратами, которые склонные к распаду под воздействием ультрафиолетового излучения. Например, офтальмологическая лекарственная форма капли ципрофлоксацина 0,3 % под воздействием ультрафиолетового света подвергается фотолизу в результате чего образуется этилендиаминовая форма, аналог ципрофлоксацина (22).
- Декарбоксилирование. Карбоновые кислоты (-COOH) имеют тенденцию к утрате диоксид углерода из карбоксильных групп при нагревании. Например, фотореакция кишечных таблеток руфлоксацина, покрытых фталатом ацетата целлюлозы, совместно с карбонатом кальция вызывает гидролиз фталатацетата целюлозы. Эта реакция освобождает уксусную кислоту, в результате взаимодействия которой с кальция карбонатом в качестве побочного продукту выделяется углекислый газ.
- рН. Совершенно ясно, что рН, особенно его крайние значения, может способствовать гидролизу ФС, когда она находится в водном растворе в ионизированном состоянии. Поэтому при необходимости производства такой лекарственной формы необходим буферный контроль.
- Упаковочные материалы. Примеси могут возникать из упаковочных материалов (например, тары и укупорочных материалов) (23).
 Рисунок 4: Примеси оланзапина, образующиеся вследствие воздействия воздуха, нагревания и процесса производства.
Рисунок 4: Примеси оланзапина, образующиеся вследствие воздействия воздуха, нагревания и процесса производства.
Так были определены две примеси оланзапина как 1 и 2 (Рисунок 4) (24). Их структуры свидетельствуют о том, что обе примеси являются продуктами распада в результате окисления тиофенового кольца оланзапина.
С точки зрения контроля качества, ускоренный распад предоставляет данные в поддержку следующего (25):
- идентификации возможных продуктов распада
- путей распада и внутренней стабильность молекулы препарата
- валидации стабильности для определения аналитических методик
- содействия разработки аналитических методик оценки стабильности
- понимания распада ФС до целесообразных продуктов.
- скрининга для возможного определения потенциальных генотоксинов.
В нормативных документах рассматриваются различные вопросы (3-6). Ключевыми из них являются следующие:
- Ускоренный распад , как правило, выполняют на одной серии материала.
- Условия ускоренного распада являются более жесткими относительно условий ускоренных исследований стабильности, например, 50 °C; ≥ 75% относительной влажности; условия освещенности превышают нормы ICH, высокий и низкий рН; и окисление.
- Испытание фотостабильности должно быть неотъемлемой частью плана исследования ускоренного распада (10).
- Продукты распада, которые никогда не образуются при проведении исследований ускоренной или долгосрочной стабильности, могут быть не выделены или их структура может быть не определена.
- Следует учитывать массовый баланс.
Некоторые вопросы не рассматриваются в нормативных документах (3-6). Ключевыми из них являются:
- Точные условия эксперимента (температура, продолжительность и степень распада)
- Дизайн эксперимента (по усмотрению заявителя).
Примеси-метаболиты
Примеси-метаболиты являются субпродуктами, которые образуются в организме после всасывания лекарственного вещества. В процессе метаболизма ФС и препарат подвергаются в организме воздействию различных ферментов, в результате чего могут образоваться примеси-метаболиты (26-34). Метаболизм лекарственных средств традиционно делится на два этапа: метаболический (например, печеночный) клиренс + фаза I и фаза ІІ метаболического процесса. Такое разделение основано на наблюдении, что лекарственное вещество сначала подвергается окислительной атаке (например, бензол окисляется до фенола) и новая веденная гидроксильная группа далее подвергается глюкуронированию (например, фенол до фенилглюкуроновой кислоты). Некоторые метаболиты в ходе развития процесса преобразовываются в примеси. Контроль таких примесей-метаболитов, образовавшихся в процессе, в конечной ФС может не требоваться, если был проведен и учтен контроль других примесей. Снижение предельных значений, следовательно, также может не понадобиться.
Примерами могут служить примеси азенапина N-оксида, азенапина десметила и ципрофлоксацина етилендиамина, которые образуются в ходе производственного процесса, а также являются примесями метаболического процесса. (см. Рисунок 5). Возникает вопрос о том, является ли необходимым ограничение таких примесей-метаболитов в конечной ФС.
Выбор аналитических методик
Разработка новых лекарственных средств требует того, чтобы на различных этапах разработки препарата получаемые аналитические данные будут значимыми и достоверными. Препарат также должен обладать превосходной стабильностью на протяжении всего своего строка годности. Для выполнения этих требований методики должны быть разработаны таким образом и обладать такой достаточной чувствительностью, чтобы определять низкие уровни примесей. Эти требования должны привести к созданию аналитических методик, которые подходят для определения следовых и ультраследовых уровней (например, субмикрограммовых) количества различных химических объектов (35-39). Существуют различные методы контроля примесей.
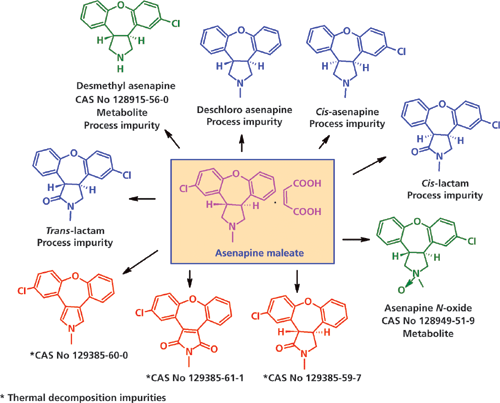
Рисунок 5: Примеси в процессе производства, примеси термического распада и метаболиты азенапина. CAS No. – Номер химической реферативной службы.
Спектроскопические методы.
Для характеризации примесей могут быть использованы различные спектроскопические методы, такие как УФ спектроскопия и спектроскопия в видимой области, ИК-спектроскопия с Фурье преобразованием, ЯМР-спектроскопия и масс-спектрометрия (МС).
Методы разделения.
Могут быть использованы различные методы разделения, в том числе тонкослойная хроматография (ТСХ), газовая хроматография (ГХ), высокоэффективная жидкостная хроматография (ВЭЖХ), капиллярный электрофорез (КЭ) и сверхкритическая жидкостная хроматография (СЖХ). Обзор этих методов приводится в литературе (39). КЭ является методом электрофореза, который часто относят к хроматографическим методам, поскольку он использует много схожих общих хроматографических требований. При помощи ТСХ с использованием различных пластин и подвижных фаз может быть разделен широкий спектр соединений. ГХ является подходящим методом для количественного определения. Он может обеспечить необходимое разрешение, селективность и простоту определения. Этот метод подходит для определения летучих органических примесей. СЖХ имеет некоторые преимущества перед ГХ в плане определения и перед ВЭЖХ с точки зрения разделения.
Комбинированные методы.
Для эффективного контроля примесей могут быть использованы следующие комбинированные методы: ГХ-МС, жидкостная хроматография (ЖХ)-МС, ЖХ-МС с диодно-матричным детектированием (ДМД), ЖХ-ЯМР, ЖХ-ЯМР-МС с ДМД и ЖХ-МС-МС.
Выделение примесей.
Часто может потребоваться выделение примесей, вследствие недоступности инструментальных методов или если требуется дальнейшее подтверждение. Для выделения примесей используются следующие методы: твердофазная экстракция, жидкостная экстракция, ускоренная экстракция растворителями, сверхкритическая жидкостная экстракция, колоночная хроматография, флэш-хроматография, ТСХ, ВЭЖХ, КЭ, и СЖХ.
Составление профиля распределения примесей.
В идеале профиль примесей должен отображать все примеси в единой форме для обеспечения контроля любых изменений в профиле. Движущими условиями для изучения профиля примесей являются обеспечение качества и нормативные требования.
Этапы для которых создаются профилей примесей.
Профиль примесей должен быть создан для ФС, процесса проверки синтеза или разработки состава, а также для конечного продукта.
Компоненты, входящие в профиль примесей.
В идеале профиль примесей должен отображать примеси, связанные с процессом синтеза, примеси, связанные с производством лекарственного препарата, продукты распада и продукты взаимодействия.
Ключевые факторы для контроля примесей в ФС.
В ходе контроля примесей в ФС важными является несколько факторов, которые изложены ниже.
Кристаллизация.
Размер кристаллов определяет не только качество, но и стабильность препарата. Во время кристаллизации должны сформироваться кристаллы высокого качества во избежание захвата незначительного количества химического вещества из маточного раствора, что, в свою очередь, приводит к распаду препарата.
Промывание отфильтрованного осадка.
Многие нежелательные химические вещества, в том числе остаточные растворители, могут быть удалены при тщательном промывании отфильтрованного осадка, что в случае неправильного выполнения может привести к задержке растворителей и примесей в осадке.
Высушивание.
Использование вакуумной сушки или сушки псевдоожиженного слоя рекомендуется в противовес лотковой сушке. Использование первой сокращает время сушки и делает её равномерной, что может быть полезным в процессе сушки лабильных лекарственных веществ.
Правильная упаковка.
При фасовке/упаковке нерасфасованного ЛС должны учитываться его характер и лабильность. Светочувствительные препараты должны быть упакованы в светозащитную упаковку. Использование непрозрачных контейнеров для глазных капель ципрофлоксацина защищает активные ингредиенты от фотораспада (22). Использование ампул с черной углеродной бумагой или с алюминиевой фольгой для эргометрина дает незначительный распад последнего (40). Также важным фактором является подбор наиболее подходящего контейнера и его укупорочного материала.
Методы производства основанные на исследовании стабильности.
Производитель нерасфасованных форм ЛС должен провести детальное исследование процесса, в том числе исследование стабильности на завершающем этапе процесса производства. Например, для производства инъекционного диклофенака натрия процесс асептической фильтрации является предпочтительным, по сравнению с автоклавированием, которое способствует появлению примесей (16).
Определения в соответствии с Фармакопеями.
Фармакопеи должны принимать меры по включению норм для примесей фармацевтических субстанций, в которых данные примеси контролируют. Это также целесообразно и для потребителей, если нормы для примесей указывают на лекарственных формах.
Заключение
В частях I, II и III этой статьи описаны виды, происхождение, причины, химия и воздействие примесей в ФС и лекарственных препаратах (1, 2). В частях I и II объяснено как, когда и почему происходит образование примесей . Эта статья, часть III, описывает примеси, связанные с распадом, процессом производства препаратов, а также примеси-метаболиты, различные аналитические методики для их идентификации и разделения, а также ключевые факторы, которые следует контролировать при производстве нерасфасованных форм ЛС.
Литература
1. K.R. Wadekar et al., Pharm. Technol. 36 (2), 46–51 (2012).
2. K.R. Wadekar et al., Pharm. Technol.36 (3), 58–70 (2012).
3. ICH, Q1A (R2) Stability Testing of New Drug Substances and Products (Nov. 2003).
4. ICH, Q3A(R) Impurities in New Drug Substances (Feb. 2003).
5. ICH, Q3B (R) Impurities in New Drug Products (Nov. 2003).
6. ICH, Q3C (R5), Impurities: Guideline for Residual Solvents (March 2011).
7. K.M. Alsante et al., Am. Pharm. Rev.4 (1), 70–78 (2001).
8. T.R. Sharp, Am. Pharm. Rev.9 (7), 84–91 (2006).
9. T.R. Sharp, Am. Pharm. Rev. 9 (3), 100–105 (2006).
10. J.A. Mollica et al., J. Pharma. Sci.67 (4), 443–465 (1978).
11. S. Ahuja, Impurities Evaluation of Pharmaceuticals (Marcel Dekker, New York, 1998)
12. L. Gu et al., Int. J. Pharm. 41 (1–2) 105–113 (1988).
13. P.V. Devarajan et al., J. Pharm. Biomed. Anal.22 (4), 679–683 (2000).
14. M.C. Damle et al., J. Adv. Sci. Res. 2 (3), 77-82 (2011).
15. K.A. Connors, G.L. Amidon, and V. J. Stella, Chemical Stability of Pharmaceuticals—A Handbook for Pharmacists (John Wiley & Sons, New York, 1986).
16. J. Roy et al., J. Pharm. Sci.90 (5) 541–544 (2001).
17. G.J.A. Walker et al., Lancet13 (2), 393–393, (1988).
18. H.V. Hogerzeil et al., British Medical Journal304 (25), 210–212 (1992).
19. J. Roy et al., Indian Drugs34 (11), 634–636 (1997).
20. J. Roy et al., Drug Dev. Ind. Pharm.20 (13), 2157–2163 (1994).
21. P.S. Rao et al., J. Pharm. Biomed. Anal.56 (2) 413–418, (2011).
22. E. Fasani et al., Photochem. Photobiol.68 (5) 666–674 (1998).
23. K. M Alsante et al., J. Pharm. Sci.93 (9) 2296-2309 (2004).
24. W. Steven et al., J. Pharm. Sci. 97 (2) 883–892 (2008).
25. S.W. Baertschi et al., Pharm. Technol.26 (2) 48–54 (2002).
26. J. Tagg et al., Biochem. Pharmacol.16 (1) 143–153 (1967).
27. F.M. Eckenrode, J. Nat. Prod. 47 (5) 882–884 (1984).
28. J.M. Bowen et al., Anal. Chem.53 (14) 239–2242 (1981).
29. M. Colvin, Clin. Pharmacokinetics4 (5) 380-394 (1979).
30. Z. H. Israili et al., J. Pharmacol. Exp. Ther. 187 (1) 138–151 (1973).
31. M.A. Schwartz. et al., Drug Metab. Dispos. 1 (1) 322–331 (1973).
32. K. Kassahun et al., Drug Metab. Dispos.1 (25) 81–93 (1996).
33. E. Stoermer et al., Drug Metab. Dispos. 10 (28) 1168–1175 (2000).
34. J. G. Slatter et al., Drug Metab. Dispos. 8 (29) 1136–1145 (2001).
35. S. Ahuja, Chromatography of Pharmaceuticals—Natural, Synthetic and Recombinant Products, ACS Symposium Series 512 (American Chemical Society, Washington, DC, 1992).
36. S. Ahuja, Trace and Ultratrace Analysis by HPLC (John Wiley & Sons, New York, 1992).
37. S. Ahuja, Chromatography and Separation Science (Elsevier, 2003).
38. S. Ahuja and S. Scypinski, Eds., Handbook of Modern Pharmaceutical Analysis (Academic Press, 2001).
39. S. Ahuja and M.Dong, Eds., Handbook of Pharmaceutical Analysis by HPLC: Volume 6 (Academic Press, 2005).
40. J. Roy et al., Indian Drugs 34 (11) 634–636 (1997).
Конструктивные дополнения и комментарии к циклу статей – приветствуются!
Приятного прочтения!






Спасибо! Очень полезно!
Спасибо за отличный цикл перевода статей! Прочитал все без отрыва и добавил в закладки.