В настоящей статье описывается концепция квалификации эксплуатации (PQ) на основе оценки рисков, которая базируется исключительно на фундаментальных требованиях. В результате из этой статьи получилось, своего рода, справочное руководство, описывающее общую методологию определения необходимого объема и количества циклов PQ. Фундаментальный принцип заключается в том, что PQ должна имитировать нормальное производство, включая проблемы, обычно встречающиеся в ходе него. Соответственно, вместо установления набора специфических параметров машины проводят испытание действий и оперативных мер, возникающих в условиях нормального производства. Описанная методология предназначена для практического применения.
В данной статье основное внимание уделяется квалификации эксплуатации процесса упаковки, взятой в качестве примера, однако на практике эта концепция может быть внедрена для всех механических процессов, поскольку все технологические характеристики являются одинаковыми.
В более долгосрочной перспективе эта методология будет предложена в качестве «Приложения C» к руководству Глобальной оперативной рабочей группы по гармонизации (GHTF) (1) (прим. переводчика: в данный момент эта группа переименована в Международный форум регулирующих органов по медицинским изделиям (IMDRF)) «Системы менеджмента качества — руководство по валидации процесса».
ВВЕДЕНИЕ
С тех пор как Управление по продуктам питания и лекарственным препаратам США выпустило в 1980-х годах первые требования к валидации фармацевтических процессов (2), и особенно в последние годы, валидация, по всей видимости, сама по себе стала становиться все более и более научной дисциплиной. Написаны книги, издаются журналы и статьи, проводятся курсы, а армия консультантов предлагает свои экспертные услуги фармацевтическим компаниям.
Часто этот подход содержит в себе философские соображения касательно более или менее конкретных заявлений, опубликованных официальными органами. Эти соображения всегда влекут за собой заявления типа «Это решать вам или вашей компании» или «Вы должны разработать обоснование для выполняемых операций … и вы тот, кто знает процесс». Эти заявления влекут за собой аналогичный ответ от «занятых в процессе» людей: «Да, мы очень хотели бы подготовить документальное подтверждение, которое предоставит высокую степень уверенности в том, что конкретный процесс будет постоянно давать продукт, который соответствует заданной спецификации и качественным характеристикам (2), но как мы можем сделать это на эксплуатационном уровне?».
Подобный сценарий проявился во многих компаниях. Стандартные операционные процедуры (СОПы), касающиеся валидации, стали все более и более философскими, отдаляясь от эксплуатационного подхода. И поскольку фармацевтические компании извлекли уроки в 1990-х годах и приняли «валидацию» как часть повседневного бизнеса, то шаблонные решения стали все более и более сложными и наполненными обоснованиями для выполнения неясных требований.
Чтобы устранить разрыв между философскими соображениями и эксплуатационными характеристиками валидации, GHTF еще в 1999 году (ред. 1) и в 2004 (ред. 2) опубликовала руководство по валидации процесса (1). Это руководство дало ответы на многие вопросы, но при этом некоторые остались и без ответа, такие как:
- Каков минимальный объем серии для квалификации эксплуатации (PQ) процессов сборки и упаковки?
Серия для PQ должна быть промышленного объема (3), однако для упаковочных операций некоторые клиенты заказывают три единицы, а другие клиенты заказывают 500 000 единиц. Следовательно, будет ли правильным выполнить квалификацию на сериях для PQ с объемом по три единицы каждая?
Тогда следует предполагать, что объем 500 000 единиц является сценарием «наихудшего случая». Но на самом деле это не так, поскольку, когда линия запущена и работает, то она выполняет одни и те же операции снова и снова, независимо от того нужно ли их выполнять для трех единиц или для 500 000 единиц. Соответственно, какие соображения и обоснования могут быть использованы для определения подходящего объема серии для циклов PQ?
- Какое минимальное количество серий необходимо для PQ?
Понятно, что «три» не является «вымышленным» ответом (4, 5), однако какие соображения и обоснования могут быть использованы для определения необходимого количества циклов PQ?
И хотя в приложении A («Статистические методы и инструменты для валидации процесса»), а также в приложении B («Пример валидации») к руководству GHTF по валидации процесса (1) очень четко и в достаточной мере описаны методологии выполнения философских требований к квалификации монтажа (IQ) и квалификации функционирования (OQ), то в секции PQ нет какой-либо методологии в отношении эксплуатации. Поэтому в настоящей статье основное внимание уделено аспектам PQ и предлагается общая методология для перевода философских определений PQ в эксплуатационные термины.
Область применения данной статьи охватывает определение и описании подхода на основе оценки рисков для выполнения квалификации эксплуатации процессов упаковки медицинских изделий и предварительно заполненных устройств. Эта статья не охватывает IQ или OQ, а только затрагивает цель этих этапов валидации процесса. Основное внимание этой статьи сконцентрировано на процессах самой упаковки, взятых в качестве примера.
ОБЛАСТЬ ПРИМЕНЕНИЯ
Область применения технологического руководства GHTF «Системы управления качеством — Руководство по валидации процесса» (1) ограничивается медицинскими изделиями, однако, как на счет сборки предварительно заполненных устройств и упаковки в целом? Предварительно заполненные устройства обычно регистрируют как лекарственные препараты, следовательно, они попадают под требования частей 210 и 211 кодекса 21 CFR FDA «Действующие требования надлежащей производственной практики к производству, обработке, упаковке и хранению лекарственных препаратов», но технологические характеристики сборки предварительно заполненных устройств и процессов упаковки больше схожи с процессами, используемыми для производства медицинских изделий, чем с фармацевтическими процессами. Поэтому, как и обсуждалось в статье Джона Шарпа (Sharp J.) «Валидации — сколько же требуется?» (7), есть ли смысл тогда адаптировать конструктивные принципы испытаний, используемые для фармацевтических процессов, к области механической обработки, где характеристики процесса отличаются? Очевидный ответ — нет. Процессы сборки и упаковки намного больше похожи на таковые процессы для медицинских изделий, нежели на фармацевтические процессы.
Поскольку процессы упаковки являются характерными механическими процессами, в ходе которых за раз обрабатываются несколько элементов в периодической последовательности (то есть часто проверяются и контролируются после каждого этапа), этот подход может быть расширен для охвата процессов сборки в целом, поскольку технологические характеристики одинаковые. Эти технологические характеристики несколько контрастируют с традиционными фармацевтическими процессами, характеризующимися химическими реакциями и операциями смешивания. Поэтому читатель должен самостоятельно решить, является ли предложенная базовая методология значимой для фармацевтических процессов.
(прим. пер. – поскольку оригинальное руководство, упоминаемое в статье, уже перенесено в архив и его достаточно сложно найти на старом сайте, я прикладываю его к этой статье, для скачивания нажмите на эту ссылку: GHTF Quality Management Systems – Process Validation Guidance.)
ВАЛИДАЦИЯ ПРОЦЕССА УПАКОВКИ
Процесс упаковки упоминается как в кодексе 21 CFR FDA, части 210 и 211 (по лекарственным препаратам) (6), так и в кодексе 21 CFR FDA, части 820 (медицинские изделия) (8). В то время как части 210 и 211 (для лекарственных препаратов) кодекса 21 CFR FDA в основном сосредоточены на информации касательно маркировки и содержимого, в части 820 кодекса 21 CFR FDA (для медицинских изделий) также рассматривается и сама упаковка.
В части 820.130 (для медицинских изделий) кодекса 21 CFR FDA указано: «Каждый производитель должен удостовериться в том, что упаковка и транспортная тара медицинского изделия спроектированы и изготовлены так, чтобы обеспечивать защиту изделия от изменения или повреждения в ходе обычных условий обработки, хранения, отгрузки и дистрибуции».
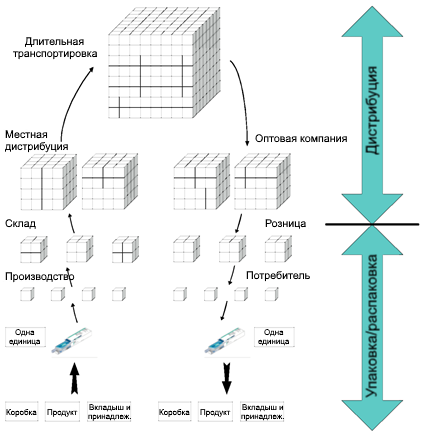
Это определение подразумевает, что для всей цепочка (упаковки) (см. рисунок 1) должно быть «подтверждение того, что она функционирует как положено».
(картинки все кликабельные, нажимайте на них, чтобы просмотреть с большей резкостью)
Основными функциями упаковки являются удержание, переноска и защита продукта. Для передачи конкретного продукта с места производства в место его использования требуется некоторый контейнер, который не только бы удерживал продукт, но и защищал его от внешних повреждений. Если продукт не предназначен для немедленного использования, то он должен сохраняться с помощью какого-то подходящего процесса. Соответственно, упаковка также будет служить барьером, изолирующим сохраняемый объект от внешнего загрязнения и порчи. Кроме того, в современном обществе упаковка используется для коммуникативных целей, таких как продвижение, объяснение, рекламирование и, особенно в случае фармацевтических препаратов, для различения содержимого. Все эти аспекты применимы к общей упаковке, но что особенно важно для фармацевтических препаратов, так это то, что они гораздо более жестко регулируются, чем остальные отрасли в целом.
На рисунке 1 проиллюстрирован жизненный цикл упаковки. Несмотря на то, что «валидация процесса упаковки» в классическом контексте применима исключительно к самому процессу упаковки (то есть к маркировке, добавлению листков-вкладышей/ИМП, помещению в картонную пачку, обёртывание и помещение в транспортную тару), тем не менее, она также охватывает и всю цепочку дистрибуции.
Следуя общей модели валидации, показанной на рисунке 2 (5), валидация процесса упаковки является суммой всех операций (то есть квалификации, сертификации и верификации), позволяющих констатировать, что вся конструкция упаковки будет работать как положено. В соответствии с моделью на рисунке 2 (5), это подразумевает, что каждый элемент во всем жизненном цикле упаковки (то есть в процессе разработки и промышленного производства) должен быть квалифицирован.
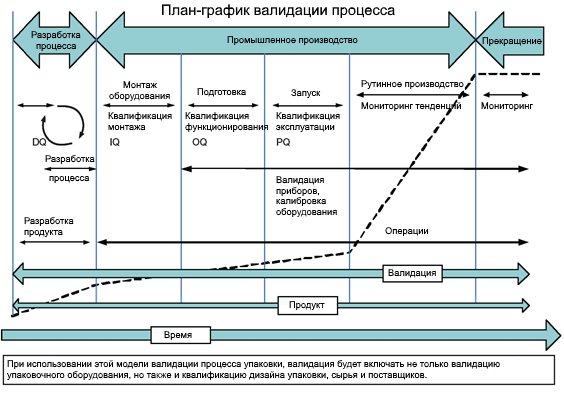
Разработка процесса — Квалификация проектной документации
Конструкция и дизайн упаковки должны быть квалифицированы. Обычно это выполняют путем проведения следующих испытаний: имитация транспортировки, стрессовая транспортировка, транспортировка в реальных условиях и использование потребителем. Эти методики испытаний в некоторой мере стандартизированы в серии документов по транспортировке Международной организации по стандартизации (ИСО) (9).
Промышленное производство — Материалы и поставщики должны быть квалифицированы
Поскольку за готовый продукт ответственность несет владелец регистрационного удостоверения, его компания также несет ответственность и за всю цепочку поставки, включая и то, что мог поставить субподрядчик. Кроме того, обычно сырье для упаковочных операций (например, этикетки продуктов, листки-вкладыши/ИМП и картонные пачки) предоставляется субподрядчиками или поставщиками, в этом случае компания несет ответственность не только за сами физические материалы, но и за напечатанную информацию. Качество и безопасность в цепочке поставки обычно обеспечиваются за счет полного набора спецификаций, определяющих правильные физические свойства и правильную информацию о данной поставке. На этапе проектирования необходимо убедиться в том, что настройка спецификации отражает правильные аспекты функциональных и маркетинговых критериев, производственных и стоимостных критериев, отношения покупателя и поставщика, а также принцип контроля качества.
Промышленное производство — Квалификация процесса упаковки
Типичная высокоскоростная упаковочная линия состоит из нескольких единиц специализированного оборудования, обычно последовательно соединенного движущимся конвейером. На рисунке 3 показана типичная упаковочная линия.
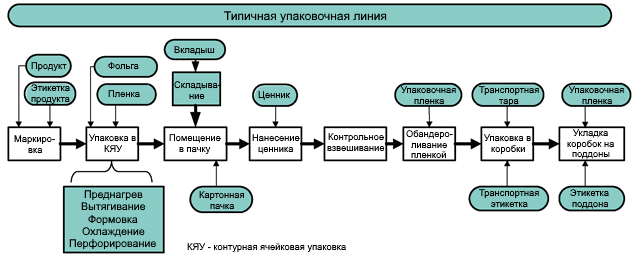
В зависимости от конкретной настройки линия может включать больше или меньше из этих основных процессов. В некоторых случаях линию делят на более автономные единицы, а в некоторых случаях отдельные процессы выполняют вручную.
Валидационные этапы
Как показано на рисунке 2, квалификацию оборудования осуществляют в ходе следующих общепринятых этапов:
- в ходе IQ проверяют правильность монтажа оборудования в соответствии с инструкциями (как правило, инструкциями производителя)
- в ходе OQ проверяют, что оборудование способно функционировать, как и ожидалось, во всех предполагаемых условиях, в том числе в условиях «наихудшего случая». Понятно, что IQ должна быть завершена до начала OQ.
- в ходе PQ проверяют, что в нормальных условиях эксплуатации оборудование стабильно дает продукцию приемлемого качества. OQ должна быть завершена до начала PQ.
- В ходе рутинного производства контролируют тенденции по ежедневным производственным данным и потребительским рыночным данным.
Несмотря на то, что IQ и OQ кажутся управляемыми, PQ часто кажется проблематичной и сложной. Об этом упоминается в различных источниках. Например, Кэрол ДеСейн (Carol DeSain) заявляет: «Она (PQ) не подразумевает бесконечного количества демонстрационных циклов с каждым параметром, установленным на минимальный или максимальный предел» (10, стр. 22). В дополнение, Джерри Ланиз (Jerry Lanese) говорит: «Сколько циклов мы должны выполнить, чтобы валидировать этот процесс? Этот вопрос задается каждый раз, когда идет обсуждение валидационного проекта. Дежурный ответ — три. Но этот ответ вводит в заблуждение …» (4, Введение). Так как же PQ можно сделать тоже управляемой?
Определение «Квалификации эксплуатации»
Следуя главному принципу валидации медицинской упаковки от Рональда Пильчика (Ronald Pilchik) (11), стратегия устранения сложности из системы, ставшей слишком сложной, состоит в следующем: начать все с чистого листа, вернуться к основам и задать следующие вопросы:
- Каковы точные основные требования, которые мы собираемся выполнить? Не больше и не меньше.
- Как мы можем перевести эти требования, пункт за пунктом, в осмысленные эксплуатационные условия?
- Как мы можем реализовать эти условия, пункт за пунктом?
Возвращаясь к фундаментальным требованиям, переводим их в эксплуатационные условия и заканчиваем рекомендациями руководства по валидации процесса GHTF.SG3.N99-10 (1). Здесь отчетливо прослеживается следующее определение квалификации (раздел 2.3):
«Квалификация эксплуатации (PQ): это установление по объективным данным того, что в предполагаемых условиях процесс стабильно дает продукт, который отвечает всем предопределенным требованиям».
Это определение переводится в следующую основную цель фазы PQ (1, раздел 5.5):
«Для реализации этого определения основная цель работ PQ заключается в демонстрации того, что процесс позволит стабильно производить приемлемую продукцию в нормальном режиме эксплуатации, а проблемы в ходе этого процесса должны имитировать условия, которые будут встречаться в реальных производственных условиях. Эти проблемы должны включать ряд условий, которые будут возникать при нормальном производстве, поскольку они определяются действиями и оперативными мерами, разрешенными в документированных стандартных операционных процедурах (обычно устанавливаемых в фазе OQ). Эти проблемы также должны быть повторены достаточное количество раз для гарантии того, что результаты являются значимыми и согласованными».
Такая интерпретация дает нам эксплуатационный смысл: во-первых, мы должны определить какие условия эксплуатации являются нормальными, а во-вторых, мы должны определить какие типичные проблемные условия мы наблюдаем при нормальном производстве. Перечень типичных влияющих факторов предлагается в документе «Системы управления качеством — Руководство по валидации процесса» (1). Перечень в руководстве может быть использован для вдохновения и перекрестной проверки, однако Джерри Ланиз (5) предлагает более систематическую и последовательную методологию для идентификации переменных, которые влияют на «нормальное производство». Предложение состоит в использовании методологии «5M» (то есть человек, материал, машина, метод и измерение [man, material, machine, method, measurement]) или диаграммы Исикавы. Объединив эти две методологии можно разработать концепцию PQ на основе оценки рисков. В этом контексте предлагаемая концепция является альтернативой «обычно используемому» методу: выполнение трех заказов на производство серии среднего объема с использованием усиленного отбора проб.
Описание концепции PQ на основе оценки рисков — Методология
Концепция PQ на основе оценки рисков, показанная на рисунке 4, отражает жизненный цикл производственного заказа в нормальных условиях и возникающие в связи с этим проблемы. Она включает следующие этапы:
- Определение нормального производства
- Определение потенциальных влияющих условий
- Обеспечение охвата всех аспектов
- Обоснованное исключение нерелевантных влияющих параметров из перечня
- Определение проблемных условий путем выполнения оценки рисков для каждого влияющего параметра
- Оценка повторения каждого проблемного параметра
- Оценка и определение «стабильного» производства
- Имитация нормального производства, включая идентифицированные проблемы
- Расчет последовательности PQ, которая имитирует нормальное производство, включая проблемы
- Определение необходимого количества циклов PQ.
1. Определение нормального производства
Нормальное производство подразумевает, что должна быть проведена имитация полного жизненного цикла производственного заказа. Жизненный цикл нормального производственного заказа включает следующие три основные этапа:
- Подготовка, которая включает: оформление заказа, получение материалов со склада, проверку материалов (готовность к производству), очистку линии и настройку формата производственной линии.
- Производство, которое включает: настройку параметров обработки, проверку калибровки, запуск процедуры, выполнение заказа, документирование производства, освобождение линии, согласование (сверку), очистку линии и внутрипроизводственный контроль (ВПК).
- Прекращение, которое включает: подготовку документации на серию, возвращение неизрасходованных материалов на склад, отбор проб для инспекции и производственный контроль вне линии (OPC).
Каждый этап в жизненном цикле будет иметь свои собственные проблемы. Параметры машины обычно идентифицируют в ходе OQ.
2. Определение потенциальных влияющих условий
Для этого должна быть организовна специальная группа, состоящая из компетентных и опытных специалистов. Для выполнения задачи могут использоваться различные методы мозгового штурма, такие как интеллектуальные карты и бумажные карточки.

3. Обеспечение охвата всех аспектов
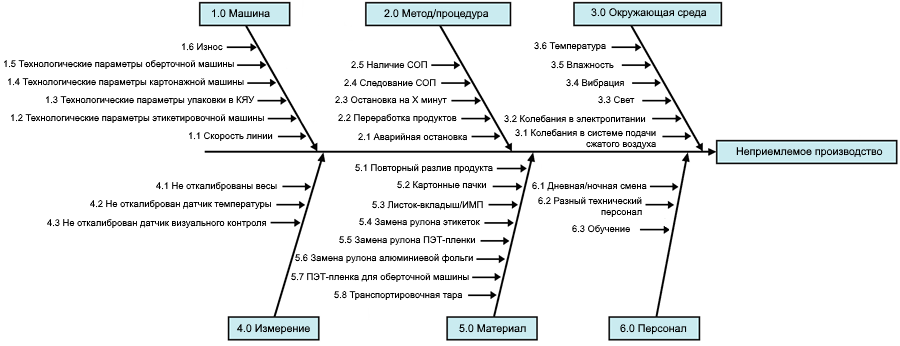
Чтобы обеспечить охват всех аспектов можно использовать диаграмму Исикавы для представления следующих объектов:
- Машина
- Метод (процедура, СОП)
- Окружающая среда
- Измерения
- Материал
- Люди (персонал).
На рисунке 5 показан пример традиционного процесса упаковки. Также можно использовать и другие структурированные методологии.
4. Исключение нерелевантных влияющих параметров
Каждый идентифицированный параметр должен быть оценен. Возможно, некоторые параметры придется принять как нерелевантные на основе очевидных аргументов и обоснования.
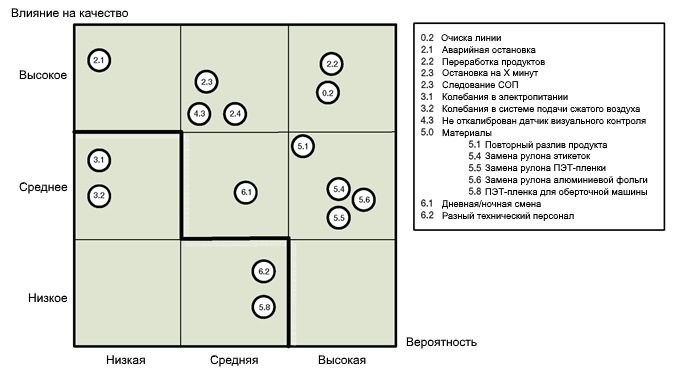
5. Определение проблемных условий путем выполнения оценки рисков
Каждый влияющий параметр оценивают с помощью матрицы «влияние на качество/вероятность риска» (см. рисунок 6). Оценка «вероятности/влияния на качество» должна основываться на опыте. Если комбинация влияния на качество и вероятности «НИЗКОЕ/НИЗКАЯ», то конкретный проблемный параметр может быть исключен из PQ. Проблемными параметрами являются те, которые расположены над диагональной линией.
6. Оценка повторения каждого проблемного параметра
Необходимо оценить, сколько раз проблемное условие должно быть повторено, чтобы гарантировать, что результаты будут значимыми и согласованными. Например, если производственный заказ обычно включает в себя картонные пачки трех разных серий, то чтобы это проблемное условие согласовывалось с нормальным производством, оно должно быть сымитировано трижды в каждом цикле PQ.
7. Оценка «стабильного» производства
При проведении PQ стабильное производство определяется как период между двумя оперативными мерами, для которого вы можете подтвердить, что предыдущая оперативная мера не повлияла на следующую.
8. Имитация нормального производства, включая идентифицированные проблемы
Если машина выполняет свою работу без вмешательства в производственную последовательность в виде каких-либо действий или оперативных мер, то она будет продолжать выполнять её без изменений снова и снова. Такова природа машины.
Проблемы могут возникнуть только при вмешательстве в производственную линию или окружающую сред путем выполнения каких-либо действий или оперативных мер. Все, что происходит в промежутке между ними, будет одинаковым. Соответственно, «промежуточные периоды» могут быть сокращены для имитации стабильного производства.
9. Расчет последовательности PQ, которая имитирует нормальное производство, включая проблемы
Имитационная последовательность может быть следующей:
- Стабильный запуск производственной линии в течение XX минут с выходом, сравнимым с нормальным выходом
- Имитация набора проблемных условий одного за другим в случайной последовательности такое количество раз, которое было оценено в пункте 6
- Стабильный запуск производственной линии между каждой имитацией во избежание взаимодействий между каждой проблемой.
В заключение, рассчитайте необходимое количество этапов так, чтобы убедиться, что весь набор проблемных условий будет охвачен в сочетании с имитационным «стабильным периодом» между каждым имитируемым проблемным условием.
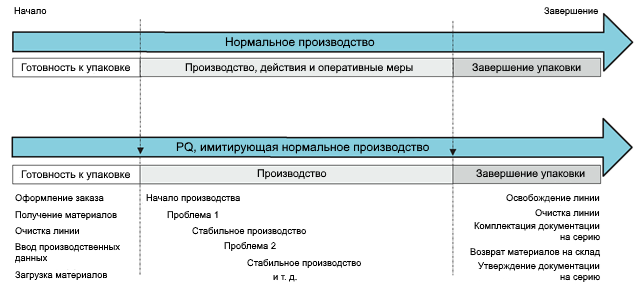
PQ, имитирующая нормальное производство, проиллюстрирована на рисунке 7. Имитация должна включать в себя те же основные элементы: подготовку, производство и прекращение производства. Однако, если нормальное производство характеризуется регулярным вмешательством в линию во время производства путем выполнения каких-либо действий и оперативных мер, то эти аспекты имитируют с помощью «сконструированных» проблем.
10. Определение необходимого количества циклов PQ
Поскольку PQ в целом должна имитировать нормальное производство, количество циклов PQ должно отражать количество основных форматов или основных вариаций, которые технические специалисты настраивают между разными заказами.
«Стабильно» является центральным словом в определении квалификации эксплуатации. Подтверждение может быть получено путем трехкратного прогона основного формата в качестве промышленного стандарта и последующего мониторинга ежедневных производственных показателей путем непрерывных пересмотров данных серии, поскольку эти действия должны быть выполнены в любом случае, так как они являются неотъемлемой частью надлежащей производственной практики (GMP) в целом и производств, регламентируемых стандартами ИСО.
После изменения, настройки и запуска нового формата оборудование будет выполнять те же самые операции снова и снова. Следовательно, нет необходимости испытывать это более одного раза. Проблемным параметром является технический специалист, который по факту будет выполнять изменения, настройку формата и запуск следующего формата. Этот параметр должен быть испытан для всех форматов.
На рисунке 8 показана предлагаемая модель цикла PQ.

Количество необходимых циклов PQ должно отражать количество возможных основных форматов.
Конфигурация основного формата будет зависеть от основных целей оборудования, обычно определяемых по конкретным промышленным параметрам. Некоторые типичные конфигурации основного формата для упаковки перечислены ниже:
- Разные продукты.
Упаковочные линии могут использоваться для упаковки нескольких продуктов при условии, что основная форма продукта и первичная упаковка одинаковые, например, таблетки одинаковой формы с разными действующими веществами (ДВ). Обычно это требует использования разных систем подачи и/или разных параметров обработки, поскольку физические и/или химические свойства будут варьироваться от ДВ к ДВ. Кроме того, проблемой могут быть барьеры, препятствующие смешиванию таблеток с разными ДВ, а также упаковка в правильные упаковочные материалы. - Разные дозируемые количества.
Линия может быть спроектирована так, чтобы фасовать и упаковывать как по 3 мл, так и по 10 мл, или она может быть спроектирована для фасовки и упаковки по 10, 100 и 500 таблеток на флакон. Обычно это потребует замены некоторых частей формата в блоке обработки этикеток. - Разное количество первичных упаковок в картонной пачке.
Часто продукт продается с разным количеством первичных упаковок на пачку, например, по 3, 5 или 10 единиц на пачку, а также часто по 1 единице в пачке. Как правило это затрагивает форматы картонажной машины, которые необходимо изменить. - Разные размеры листка-вкладыша/ИМП, помещаемых в упаковку вместе с продуктом.
Поскольку разные страны имеют разные требования к содержанию, а в ряде стран требуется, чтобы текст листка-вкладыша/ИМП был напечатан на всех официальных языках (или в случае некоторых стран на трех языках), очевидно, что это будет влиять на размер листка-вкладыша/ИМП. - Разная конфигурация упаковки.
Поскольку в некоторых странах реклама разрешена, а в других запрещена, то часто бывает так, что лучше иметь возможность добавить в упаковку больше или меньше информационных, или рекламных материалов.
ВЫВОД
В настоящей статье разработана концепция PQ на основе оценки рисков, которая базируется на фундаментальных требованиях. В результате получилось, своего рода, справочное руководство, описывающее методологию определения настроек для циклов PQ, которые имитируют нормальное производство эксплуатационным способом, включая объем серии для PQ и количество циклов PQ.
Методология сосредоточена на характеристиках нормального производства и связанных с ним проблем. Соответственно, вместо установления набора специфических параметров машины проводят испытание выполняемых действий и оперативных мер в связке с нормальным производством.
Основное внимание в данной статье уделяется квалификации эксплуатации процесса упаковки, в качестве примера, однако по сути эта концепция может быть внедрена для всех механических процессов, поскольку все технологические характеристики являются одинаковыми.
В долгосрочной перспективе эта методология будет предложена в качестве приложения C к руководству по валидации процесса (1).
СПИСОК АББРЕВИАТУР В СТАТЬЕ
| ДВ | Действующее вещество |
| FDA | Управление по продуктам питания и лекарственным препаратам США |
| GMP | Надлежащая производственная практика |
| ВПК | Внутрипроизводственный контроль |
| IQ | Квалификация монтажа |
| ИСО | Международная организация по стандартизации |
| OPC | Производственный контроль вне линии |
| OQ | Квалификация функционирования |
| PQ | Квалификация эксплуатации |
| СОП | Стандартная операционная процедура |
| КЯУ | Контурная ячейковая упаковка |
| ИМП | Инструкция по медицинскому применению |
СПИСОК ИСТОЧНИКОВ
Авторы:
Steen Howaldt Christiansen
Работает руководителем проектов в компании Ново-Нордиск А/С, Дания — отдел готовой продукции для лечения диабета / технологической разработки / готовой продукции.
и
Jesper Bønnelycke Torp Jensen
Работает инженером в компании Ново-Нордиск А/С, Дания — отдел готовой продукции для лечения диабета / технологической разработки / готовой продукции.
По материалам IVT Network