
Вы на упаковке были когда-нибудь?
Задавались вопросом как отличить упаковку от пусконаладки?
Наблюдали ли за спором контролера и налачдика: «Да она такой родилась», с привлечением всех возможных сотрудников?
Задавались ли проблемой расчета реальной производительности, расчетом числа остановок (времени простоя)?
Обращались ли к Господу с вопросом «Как в этом бардаке удается таки получать результат?»
И если вам приходилось быть третейским судьей в споре ОКК, производства и службы главного инженера по поводу брака, невыполнения плана, неработоспособности чего-либо, тогда мы идем к Вам и расскажем о попытках и подходах к валидации процесса упаковки.Тезис.
На практике не представляется реальным, особенно на новой машине или линии (ну а так же на старой) безошибочное производство без мелких сбоев, отклонений или сигналов тревоги.
Производительность машины исключает возможность текущего контроля со стороны человека.
Таким образом к валидации подходим не со стороны проверки работоспособности оборудования а со стороны проверки качества готового продукта. Для этого создаем классификацию дефектов.
Скорее всего она у вас есть в сопе по рекламациям и отзыве продукта с рыка. У хофмман ля рош она была там. Там возможные дефекты разбиты на классы опасности. Но беда в том что использовать ее для валидации процессов упаковки нельзя так как там перечислены проблемы которые не могут возникнуть или быть выявлены на данном этапе производства. Поэтому таблицу необходимо переработать в этом ключе.
Мы используем следующую классификацию:
Классификация дефектов
Классификация дефектов приведена в соответствии с СОП «Порядок работы с рекламациями». Дефектом считается любое несоответствие единицы продукции требованиям спецификации. В данном разделе приведены дефекты выявляемые контролером на участке упаковки. Дефекты разбиваются на следующие классы:
Критический класс 1 (К1)
Дефекты/претензии, которые потенциально являются опасными для жизни, или могут обусловить серьезный риск для здоровья
- Перепутывание полупродукта. Ангро не соответствует маркировке блистера, либо вторичной упаковки.
- Отсутствие печати (№ серии, срока годности)
- Не герметичность блистера.
Критический класс 2 (К2)
Дефекты/претензии, которые могут вызвать заболевание или привести к ненадлежащему лечению, но не относящиеся к Классу 1
- Отсутствующая или недостоверная информация в инструкции по медицинскому применению
- Смешивание продуктов в коробе/контейнере (“сортовая примесь”).
- Небезопасная/дефектная упаковка, обуславливающая серьезные медицинские последствия.
- Загрязнение (например, бактериальная порча, грязь или мусор или материал инородных макрочастиц). Загрязнение продукта, внутренней поверхности блистера.
Существенный (С) (Значительный) дефект
Дефекты/претензии, которые могут привести к отсутствию заявленных результатов при использовании единицы продукции, содержащей это несоответствие
- Расколотые таблетки/капсулы.
- Органолептическая проблема (например, нетипичный горький вкус)
- Неверное заполнение / вложение. Пустой или неполный (блистер, упаковка из фольги или флакон).
- Загрязнение посторонними неопасными веществами
- Нарушение значащего текста / функциональные дефекты
- Нарушение первичной упаковки (сборка контейнера / крышки, нарушение целостности пакета или блистера) – дефект, влияющий на вложенный продукт или нарушающий функциональные качества упаковки
Незначительный (Н)
Дефекты/претензии, которые не могут обусловить риск для здоровья.
- Повреждение контейнера, пакета, блистера, не затрагивающее продукта / не приводящее к функциональным нарушениям
- Закрывание: неправильно поставленная / поврежденная крышка, не затрагивающее продукта / не приводящее к функциональным нарушениям
- Внешнее загрязнение посторонними неопасными веществами
- Этикетка: неудовлетворительное приклеивание / перекос
- Повреждение компонента, не влияющее на текст или функциональность (Повреждение этикетки, не влияющее на понимание текста / функциональность)
Как я понимаю – идеология такая – опасно – вредно – не красиво
Далее воспользуемся документом ГОСТ Р ИСО 2859-1-2007 Статистические методы. Процедуры выборочного контроля по альтернативному признаку. Часть 1. и установим допустимые уровни качества.
Допустимые уровни качества (AQL) ( Acceptable Quality Level) – это максимальное количество несоответствий на сотню единиц продукции, т.е. максимально допустимый процент несоответствий, который принят в качестве удовлетворительного.
На предприятии приняты следующие уровни качества:
| Уровень | AQL |
| Критический класс 1 (К1) | 0,10 % |
| Критический класс 2 (К2) | 0,65 % |
| Существенный (С) (Значительный) | 1,5 % |
| Незначительный (Н) | 6,5 % |
Цифры взяты из сопа хоффман ля рош и одного обучающего пособия.
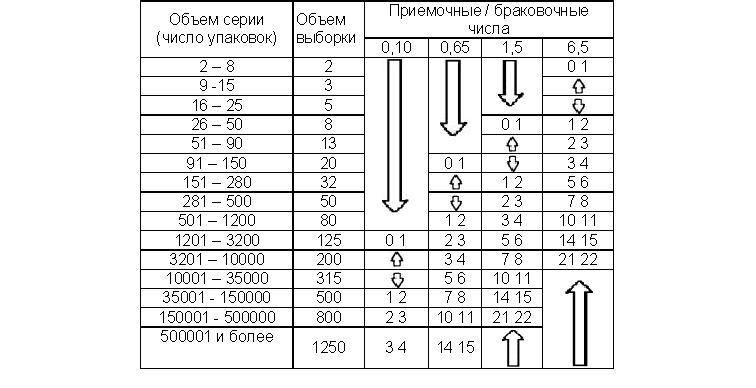
Далее, на основе Таблицы II-А (Нормальный).- Одноступенчатые выборочные планы при нормальном контроле. Составляем план выборочного контроля.
 В этом госте указан ряд процедур перехода с одного плана на другой, но на практике это не осуществимо.
В этом госте указан ряд процедур перехода с одного плана на другой, но на практике это не осуществимо.
Теперь самое интересное. Как организовать пробоотбор. Связь пространства и времени.
4.1. Определить объем выборки по объему серии по приведенной выше таблице.
4.2. Рассчитать предполагаемое время производства разделив объем серии на производительность линии.
4.3. Рассчитать необходимое число подходов к линии каждый час и необходимое число образцов продукции, которое нужно отобрать во время каждого подхода.
4.4. Отобрать необходимое количество образцов готовой продукции.
4.5. Провести проверку образцов в соответствии с приведенной классификацией. Бракованная продукция складывается в короб с надписью «Брак» годная продукция возвращается на линию.
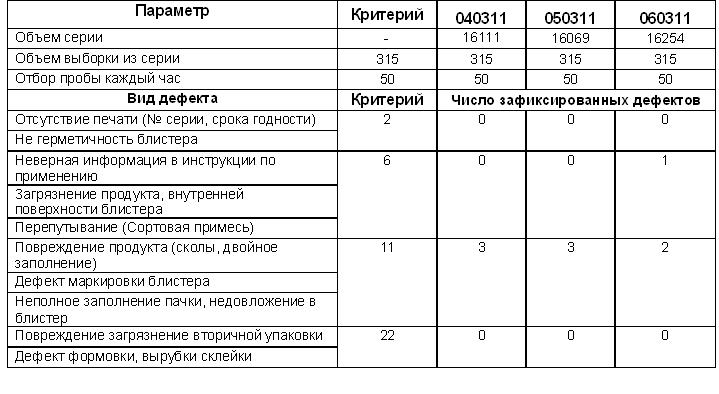
4.4. Записать число зафиксированных дефектов в заполняемую форму Приложение №1 настоящего СОПа.
4.5. Произвести расчет числа зафиксированных дефектов и сравнить их с приемочными / браковочными числами. Если число дефектов окажется выше приведенного в таблице браковочного числа, то серия бракуется. Собственно заполняемая форма.
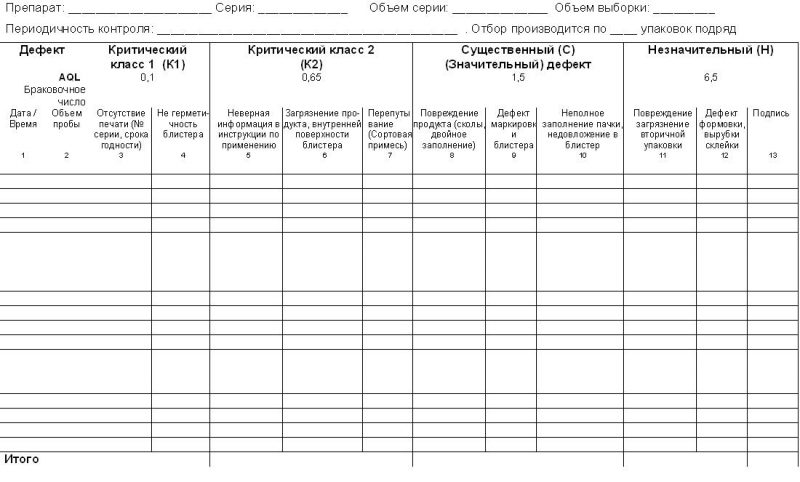
Результаты реально проведенной валидации процесса упаковки
Контролируемые параметры и критерии оценки.
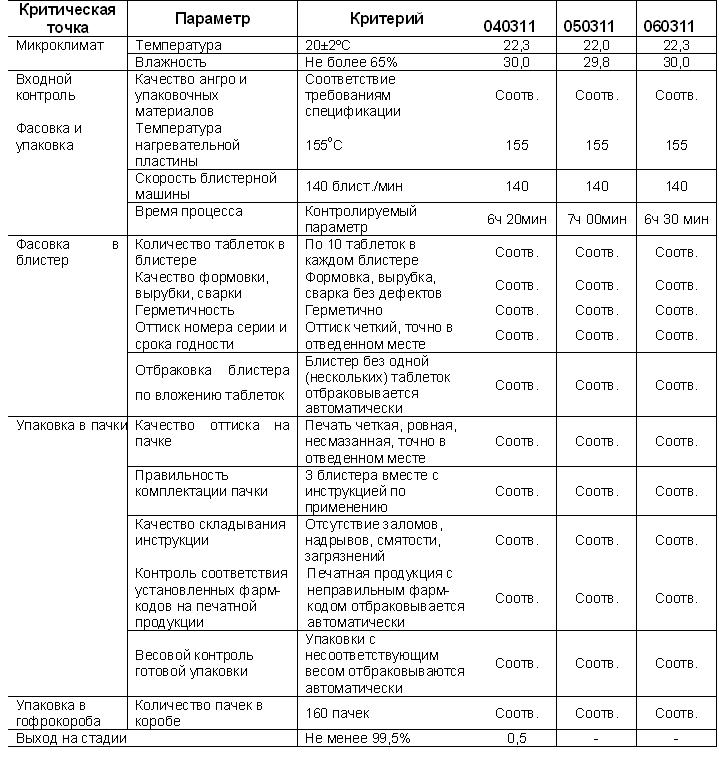 Периодичность контроля
Периодичность контроля
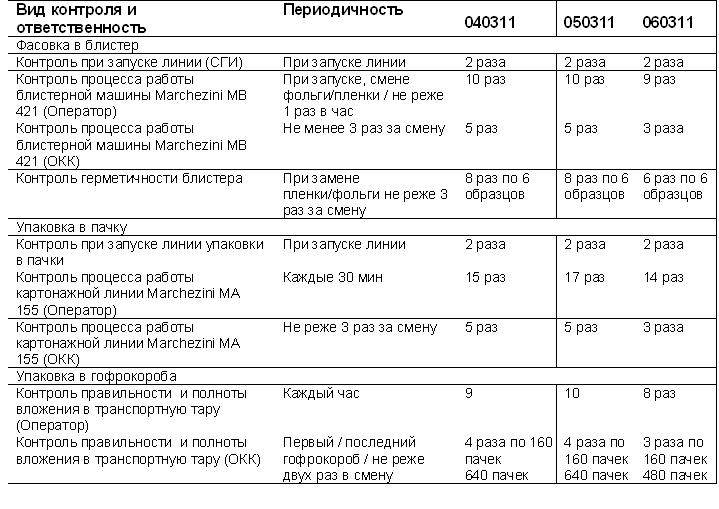 Результаты валидации
Результаты валидации
Теперь самое интересное и главный вопрос.
LethalityFactor
2. Для случая упаковки: GMP 5.54 “образцы, отобранные с упаковочной линии, нельзя возвращать назад”
Открываем ГОСТ Р 52249-2009 книжку оранжевого цвета и ищем пункт 5.54. Ан нет такого. 5.53 есть 5.55 есть, а 5.54 нет. Наверное это опечатка по Фрейду.
Тем не менее в пункте 5.53 черным по белому написано, что образцы, отобранные с линии повторно на линию не возвращают.
Тем не менее в таблице “периодичность контроля” указаны количества упаковок отбираемых с линии по документам. В реальности в разы выше. Плюс просматривается первый и последний короб контролерами ОКК.
Производственникам мой вопрос понятен?
Как всегда рад любым комментариям.






Какие еще риски могут быть вызваны непосредственно при валидации упаковочной линии? Например, PQ не вовремя, нету 3 лота в наличии, само помещение не было квалифицированно к сроку, транспортировка с завода изготовителя таблеток до упаковочной станции, какие еще?
Цитировал в редакции украинского руководства по GMP, собственно можно сравнить с оригиналом европейской версии
В части объёма выборки – Принципиально для себя ответить на вопрос, с какой целью проводится пробоотбор – с целью валидации или текущего контроля? Если с целью валидации, то всё понятно, если с целью текущего контроля – то возникает ряд встречных вопросов – однородность содержания при операции смешивания (таблетмасса) в скольки точках контролируем? если в 1-2, то нерепрезентативная выборка (как пример), или на стадии таблетирования – сколько образцов отбирается – тоже по приведённой в ГОСТ Р ИСО 2859-1-2007 схеме (тоже пример)? Примеры по каждой стадии можно приводить и другие, например по контролю качества эффективности очистки – в скольки точках пробы берёте? Смысл в том, что если процесс валидирован, то объём рутинного контроля может быть снижен, и речь уже не идёт о строго математически доказуемой репрезентативности – это философия.
Есть контакт первый валидаторщик клюнул!!! 😀 😀 😀
Странная закономерность – интересные вопросы задаются в каникулы.
Просьба, старайтесь вопросы задавать более явно а то я могу не уловить что именно вам интересно.
Это хорошо. Значит до росли до уровня начальника производства или директора по качеству.
Безусловно. Взять дот или два и завоевать Германию.
В соответствии с процедурой контроля изменений, т.е. при замене производителя субстанции, технологии изготовления (смена прямого прессования на влажную грануляцию) и т.д. Перерегистрация препарата.
Может быть с последующей очисткой а не валидацией? От валидации оборудование чистым не становится, насколько я понимаю.
– мани 😀 как сказал один возовский инспектор. Попробуйте написать что вы поняли из моего поста про валидацию очистки а там будет видно чем вам помочь. У нас используется: СОП по расчету критериев приемлемости. Протокол валидации очистки конкретного препарата с указанием списка оборудования подлежащего валидации, ответственными и т.д. и к нему приложение в виде протокола отбора проб где указаны точки пробоотбора на каждую единицу оборудования.
Валидация штука серьезная и затратная. Если вы начальник производства то подумайте сами о последствиях использования нестабильной технологии, а если просто специалист по валидации то почему вас это волнует?
В данном случае для ответа недостаточно данных. У нас загрузка на V-миксер до 150 кг. В регламенте серия 200 кг состоящая из 2-х загрузок по 100 кг. В зависимости от заявки на производство мы можем выпустить серию на 50, 100 и 200 кг и по поводу валидации не заморачиваемся. Были прецеденты когда валидация производилась по загрузкам. т.е. на 1.5 сериях. Т.е. думайте сами о необходимости подтверждения стабильности и воспроизводимости технологии.
ЗЫ. Проблемы валидации на постсоветском пространстве лежат в плоскостях психологии взаимоотношений (перекладывание ответственности), организации и ведения бизнеса (продажи первичны производство вторично) и отсутствия понимания необходимости проведения (понятно что это написано в джиэмпях, но зачем это действительно надо). Так что давайте общаться дальше.
Меня вот что интересует:
1. Какие критерии оценки для процесса и для самой упаковки: количественные и качественные?2. Какие стат. методы используются для оценки приемлемости этих критериев?
3. В каких случаях можно обойтись и без стат. обработки?
Спасибо!
Уважаемый Kirillcheg, спасибо большое за посты и желание ответить на вопросы. Мои наболевшие вопросы относятся не столько к указанным темам, сколько к процессу в целом валидации производства.
1. На производстве производится более 40 наименований в таблеточной лекарственной форме. Каждое наименование должно быть валидировано: валидационный мастер план, протокол валидации должны быть оформлены соответствующе.
А. Какими критериями руководствуются в выборе периода ревалидации, описанного в VMP?
Б. В производстве разных препаратов применяется одно и тоже оборудование с последующей валидацией очистки. Как выглядит документация по валидации очистки оборудования? (пример)
В. Нужно ли проводить валидацию при масштабировании выпускаемых серий. Например, серия составляла 175000 таблеток, выпуск серии увеличивается до 350 000 таблеток. Достаточно ли заложить стабильность масштабированных серий, Batch manufacturing records (BMR) , профили растворения? Всегда ли профили растворения достаточны при масштабировании?